CHAPTER 3 Molecules: Structures and Dynamics
This chapter describes the properties of water, proteins, nucleic acids, and carbohydrates as they pertain to cell biology. Chapter 7 covers lipids in the context of biological membranes.
Water
Water is so familiar that its role in cell biology and its fascinating properties tend to be neglected. Water is the most abundant and important molecule in cells and tissues. Humans are about two thirds water. Water is not only the solvent for virtually all cellular compounds but also a reactant or product in thousands of biochemical reactions catalyzed by enzymes, including the synthesis and degradation of proteins and nucleic acids and the synthesis and hydrolysis of adenosine triphosphate (ATP), to name a few examples. Water is also an important determinant of biological structure, as lipid bilayers, folded proteins, and macromolecular assemblies are all stabilized by the hydrophobic effect derived from the exclusion of water from nonpolar surfaces (see Fig. 4-5). Additionally, water forms hydrogen bonds with polar groups of many cellular constituents ranging in size from small metabolites to large proteins. It also associates with small inorganic ions.
Physical chemists are still trying to understand water, one of the most complex liquids. The molecule is roughly tetrahedral in shape (Fig. 3-1A), with two hydrogen bond donors and two hydrogen bond acceptors. The electronegative oxygen withdraws the electrons from the O—H covalent bonds, leaving a partial positive charge on the hydrogens and a partial negative charge on the oxygen. Hydrogen bonds between water molecules are partly electrostatic because of the charge separation (induced dipole) but also have some covalent character, owing to overlap of the electron orbitals. The strength of hydrogen bonds depends on their orientation, being strongest along the lines of tetrahedral orbitals. One can think of oxygens of two water molecules sharing a hydrogen-bonded hydrogen. Given two hydrogen bond donors and acceptors, water can be fully hydrogen-bonded, as it is in ice (Fig. 3-1C). Crystalline water in ice has a well-defined structure with a complete set of tetragonal hydrogen bonds and a remarkable amount (35%) of unoccupied space (Fig. 3-1D).
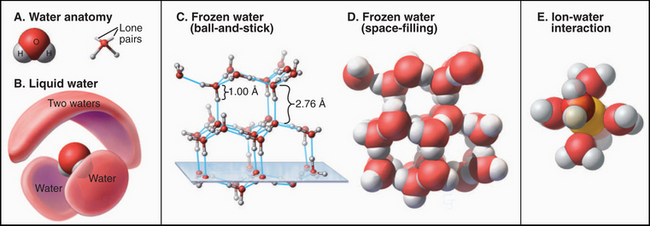
(D–E, From www.nyu.edu/pages/mathmol/library/water, Project MathMol Scientific Visualization Lab, New York University. See “ice.pdb” and “waterbox.pdb.”)
Neither theoretical calculations nor physical observations of liquid water have revealed a consistent picture of its organization. When ice melts, the volume decreases by only about 10%, so liquid water has considerable empty space too. The heat required to melt ice is a small fraction (15%) of the heat required to convert ice to a gas, in which all the hydrogen bonds are lost. Because the heat of melting reflects the number of bonds broken, liquid water must retain most of the hydrogen bonds that stabilize ice. These hydrogen bonds create a continuous, three-dimensional network of water molecules connected at their tetrahedral vertices, allowing water to remain a liquid at a higher temperature than is the case for a similar molecule, ammonia. On the other hand, because liquid water does not have a well-defined, long-range structure, it must be very heterogeneous and dynamic, with rapidly fluctuating regions of local order and disorder. This incomplete picture of water structure limits our ability to understand macromolecular interactions in an aqueous environment.
The properties of water have profound effects on all other molecules in the cell. For example, ions organize shells of water around themselves that compete effectively with other ions with which they might interact electrostatically (Fig. 3-1E). This shell of water travels with the ions, governing the size of pores that they can penetrate. Similarly, hydrogen bonding with water strongly competes with the hydrogen bonding that occurs between solutes, including macromolecules. By contrast, water does not interact as favorably with nonpolar molecules as it does with itself, so the solubility of nonpolar molecules in water is low, and they tend to aggregate to reduce their surface area in contact with water. Such nonpolar interactions are energeti-cally favorable because they reduce unfavorable interactions of nonpolar groups with water and increase favorable interactions of water molecules with each other. This is called the hydrophobic effect (see Fig. 4-5). These interactions of water dominate the behavior of solute molecules in an aqueous environment, where they influence the assembly of proteins, lipids, and nucleic acids into the structures that they assume in the cell. On the other hand, strategically placed water molecules can bridge two macromolecules in functional assemblies.
Proteins
Proteins consist of one or more linear polymers called polypeptides, which consist of various combinations of 20 different amino acids (Figs. 3-2 and 3-3) linked together by peptide bonds (Fig. 3-4). When linked in polypeptides, amino acids are referred to as residues. The sequence of amino acids in each type of polypeptide is unique. It is specified by the gene encoding the protein and is read out precisely during protein synthesis (see Fig. 18-8). The polypeptides of proteins with more than one chain are usually synthesized separately. However, in some cases, a single chain is divided into pieces by cleavage after synthesis.
Polypeptides range widely in length. Small peptide hormones, such as oxytocin, consist of as few as nine residues, while the giant structural protein titin (see Fig. 39-7) has more than 25,000 residues. Most cellular proteins fall in the range of 100 to 1000 residues. Without stabilization by disulfide bonds or bound metal ions, about 40 residues are required for a polypeptide to adopt a stable three-dimensional structure in water.
The sequence of amino acids in a polypeptide can be determined chemically by removing one amino acid at a time from the amino terminus and identifying the product. This procedure, called Edman degradation, can be repeated about 50 times before declining yields limit progress. Longer polypeptides can be divided into fragments of fewer than 50 amino acids by chemical or enzymatic cleavage, after which they are purified and sequenced separately. Even easier, one can sequence the gene or a complementary DNA (cDNA) copy of the messenger RNA for the protein (Fig. 3-16) and use the genetic code to infer the amino acid sequence. This approach misses posttranslational modifications (Fig. 3-3). Analysis of protein fragments by mass spectrometry can be used to sequence even tiny quantities of proteins.
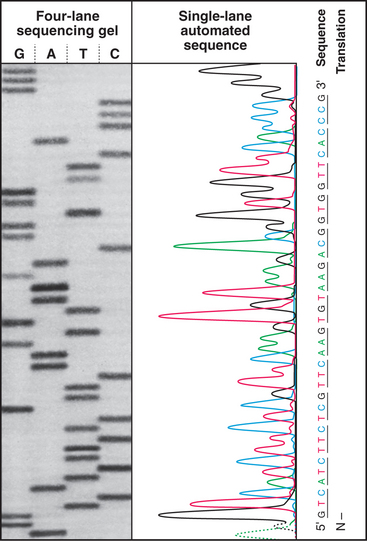
Figure 3-16 The sequence of a purified fragment of DNA is rapidly determined by in vitro synthesis (see Fig. 42-1) using the four deoxynucleoside triphosphates plus a small fraction of one dideoxynucleoside triphosphate. The random incorporation of the dideoxy residue terminates a few of the growing DNA molecules every time that base appears in the sequence. The reaction is run separately with each dideoxynucleotide, and fragments are separated according to size by gel electrophoresis (see Fig. 6-5), with the shortest fragments at the bottom. A radioactive label makes the fragments visible when exposed to an X-ray film. The sequence is read from the bottom as indicated. An automated method uses four different fluorescent dideoxynucleotides to mark the end of the fragments and electronic detectors to read the sequence.
(Based on original data from W-L. Lee, Salk Institute for Biological Studies, San Diego, California.)
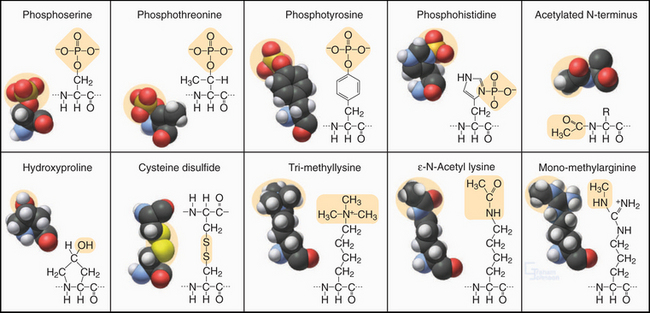
Figure 3-3 modified amino acids. Protein kinases add a phosphate group to serine, threonine, tyrosine, histidine, and aspartic acid (not shown). Other enzymes add one or more methyl groups to lysine, arginine, or histidine (not shown); a hydroxyl group to proline; or an acetate to the N-terminus of many proteins. The reducing environment of the cytoplasm minimizes the formation of disulfide bonds, but under oxidizing conditions within the membrane compartments of the secretory pathway (see Chapter 21), intramolecular or intermolecular disulfide (S—S) bonds form between adjacent cysteine residues.
Properties of Amino Acids
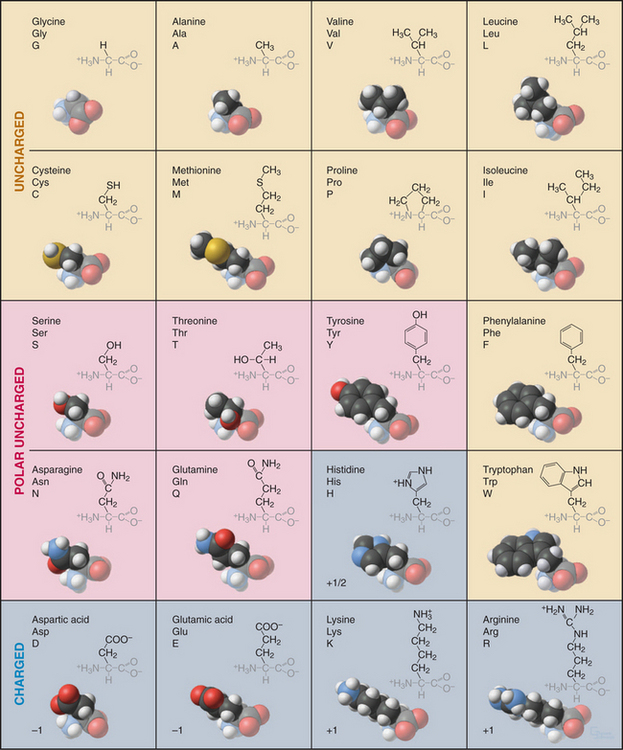
Every student of cell biology should know the chemical structures of the amino acids used in proteins (Fig. 3-2). Without these structures in mind, reading the literature and this book is like spelling without knowledge of the alphabet. In addition to their full names, amino acids are frequently designated by three-letter or single-letter abbreviations.
All but one of the 20 amino acids commonly used in proteins consist of an amino group, bonded to the α-carbon, bonded to a carboxyl group. Proline is a variation on this theme with a cyclic side chain bonded back to the nitrogen to form an imino group. Both the amino group (pK > 9) and carboxyl group (pK = ∼4) are partially ionized under physiological conditions. With the exception of glycine, all amino acids have a β-carbon and a proton bonded to the α-carbon. (Glycine has a second proton instead.) This makes the α-carbon an asymmetrical center with two possible configurations. The l-isomers are used almost exclusively in living systems. Compared with natural proteins, proteins constructed artificially from d-amino acids have mirror-image structures and properties.
Enzymes modify many amino acids after their incorporation into polypeptides. These posttranslational modifications have both structural and regulatory functions (Fig. 3-3). These modifications are referred to many times in this book, especially reversible phosphorylation of amino acid side chains, the most common regulatory reaction in biochemistry (see Fig. 25-1). Methylated and acetylated lysines are important for chromatin regulation in the nucleus (see Fig. 13-3). Whole proteins such as ubiquitin or SUMO can be attached through isopeptide bonds to lysine e-amino groups to act as signals for degradation (see Fig. 23-8) or endocytosis (see Fig. 22-16).
This repertoire of amino acids is sufficient to construct millions of different proteins, each with different capacities for interacting with other cellular constituents. This is possible because each protein has a unique three-dimensional structure (Fig. 3-5), each displaying the relatively modest variety of functional groups in a different way on its surface.
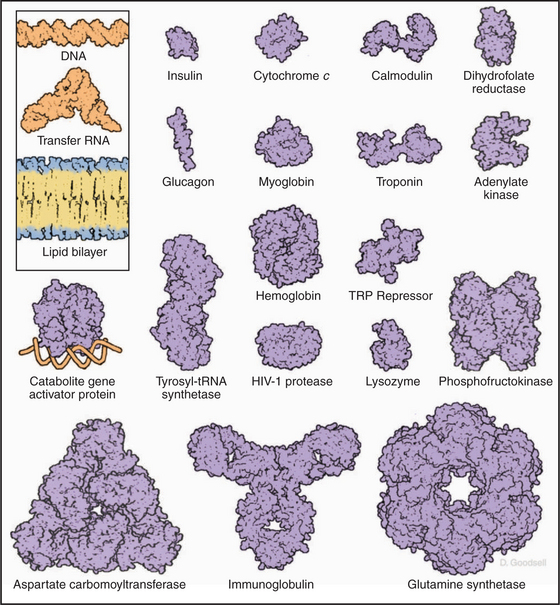
Architecture of Proteins
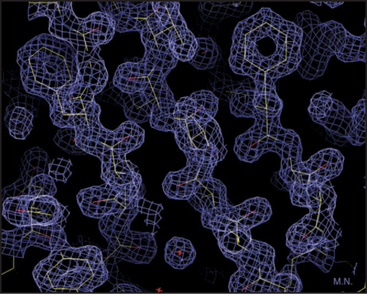
Our knowledge of protein structure is based largely on X-ray diffraction studies of protein crystals or nuclear magnetic resonance (NMR) spectroscopy studies of small proteins in solution. These methods provide pictures showing the arrangement of the atoms in space. X-ray diffraction requires three-dimensional crystals of the protein and yields a three-dimensional contour map showing the density of electrons in the molecule (Fig. 3-6). In favorable cases, all the atoms except hydrogens are clearly resolved, along with water molecules occupying fixed positions in and around the protein. NMR requires concentrated solutions of protein and reveals distances between particular protons. Given enough distance constraints, it is possible to calculate the unique protein fold that is consistent with these spacings. In a few cases, electron microscopy of two-dimensional crystals has revealed atomic structures (see Figs. 7-8B and 34-5).
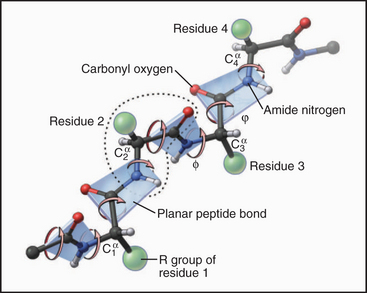
Each amino acid residue contributes three atoms to the polypeptide backbone: the nitrogen from the amino group, the α-carbon, and the carbonyl carbon from the carboxyl group. The peptide bond linking the amino acids together is formed by dehydration synthesis (see Fig. 17-10), a common chemical reaction in biological systems. Water is removed in the form of a hydroxyl from the carboxyl group of one amino acid and a proton from the amino group of the next amino acid in the polymer. Ribosomes catalyze this reaction in cells. Chemical synthesis can achieve the same result in the laboratory. The peptide bond nitrogen has an (amide) proton, and the carbon has a double-bonded (carbonyl) oxygen. The amide proton is an excellent hydrogen bond donor, whereas the carbonyl oxygen is an excellent hydrogen bond acceptor.
The peptide bond has some characteristics of a double bond, owing to resonance of the electrons, and is relatively rigid and planar. The bonds on either side of the α-carbon can rotate through 360 degrees, although a relatively narrow range of bond angles is highly favored. Steric hindrance between the β-carbon (on all the amino acids but glycine) and the α-carbon of the adjacent residue favors a trans configuration in which the side chains alternate from one side of the polymer to the other (Fig. 3-4). Folded proteins generally use a limited range of rotational angles to avoid steric collisions of atoms along the backbone. Glycine without a β-carbon is free to assume a wider range of configurations and is useful for making tight turns in folded proteins.
Folding of Polypeptides
The three-dimensional structure of a protein is determined solely by the sequence of amino acids in the polypeptide chain. This was established by reversibly unfolding and refolding proteins in a test tube. Many, but not all, proteins that are unfolded by harsh treatments (high concentrations of urea or extremes of pH) will refold to regain full activity when returned to physiological conditions. Although many proteins are flexible enough to undergo conformational changes (see later discussion), polypeptides rarely fold into more than one final stable structure. Exceptions with medical importance are prions and amyloid (Box 3-1).
BOX 3-1 Protein Misfolding in Amyloid Diseases
Given that amyloid fibrils form spontaneously and are exceptionally stable, it is not surprising that functional amyloids exist in organisms ranging from bacteria to humans. For example, formation of the pigment granules responsible for skin color depends on a proteolytic fragment of a lysosomal membrane protein that forms amyloid fibrils as a scaffold from melanin pigments. Budding yeast has a number of proteins that can either assume their “native” fold or assemble into amyloid fibrils. The native fold of the protein Sup35p serves as a translation termination factor that stops protein synthesis at the stop codon (see Fig. 17-8). Rarely, Sup35p misfolds and assembles into an amyloid fibril. These fibrils sequester all the Sup35p in fibrils, where it is inactive. The faulty translation termination that occurs in its absence has diverse consequences that are inherited like prions from one generation of yeast to the next.
The following factors influence protein folding:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


