FIGURE 9.1 DNA molecule. The double helix of the DNA is shown along with details of how the bases, sugars, and phosphates connect to form the structure of the molecule. DNA is a double-stranded molecule twisted into a helix (think of a spiral staircase). Each spiraling strand, composed of a sugar–phosphate backbone and attached bases, is connected to a complementary strand by noncovalent hydrogen bonding between paired bases. The bases are adenine (A), thymine (T), cytosine (C), and guanine (G). A and T are connected by two hydrogen bonds. G and C are connected by three hydrogen bonds.
Under physiologic conditions, the helical structure of double-stranded DNA (dsDNA) is stable due to the numerous, although weak, hydrogen bonds between base pairs and the hydrophobic interaction between the bases in the center of the helix. However, the weak bonds can be broken in vitro by changing environmental conditions; the strands are denatured and separate from each other. Once denatured, the negative charge of each strand causes the strands to repel each other. The two complementary strands can be reassociated or reannealed if the conditions change and favor this process. The renaturation will follow the rules of base pairing so the original DNA molecule is recovered.
RNA is also present in human cells and is chemically similar to DNA. RNA differs from DNA in three ways: (1) ribose replaces deoxyribose as the sugar; (2) uracil replaces thymine as a purine base; and (3) RNA is single stranded. DNA and RNA work together to synthesize proteins. Genomic dsDNA is enzymatically split into its two strands, one of which serves as the template for the synthesis of complementary messenger RNA (mRNA). As mRNA is released from the template DNA, the DNA strands reanneal. The mRNA specifies the amino acid to be added to the peptide chain by transfer RNA, which transports the amino acid to the ribosome, where peptide chain elongates.
This discussion of protein synthesis highlights that physiologically, DNA is routinely denatured, binds to RNA, and reanneals to reestablish the original DNA, always following base pair rules. These processes form the foundation of nucleic acid hybridization assays in which complementary strands of nucleic acid from unrelated sources bind together to form a hybrid or duplex. Molecular testing in the clinical laboratory consists of two major areas: (1) the use of DNA probes to directly detect or characterize a specific target and (2) the use of nucleic acid amplification technologies to detect or characterize a specific target DNA or RNA. Procedures that use probes include solid-phase assays (capture hybridization and Southern and Northern blotting), solution-based assays (protection assays and hybrid capture assays), and in situ hybridization assays. Amplification procedures include nucleic acid amplification (polymerase chain reaction [PCR], nucleic acid–based sequence amplification, transcription-mediated amplification [TMA], and strand displacement amplification [SDA]), probe amplification (ligase chain reaction [LCR]), and signal amplification (branched chain DNA assay).
Nucleic Acid Extraction
Nucleic acid extraction requires the release of DNA or RNA from the cell followed by its purification and quantitation. Nucleic acid can be extracted from a variety of specimens that include whole blood, serum, plasma, bone marrow, cerebrospinal fluid, urine, cultured cells, amniotic fluid, and paraffin-embedded tissue.1,2 The extracted DNA or RNA needs to be free of each other and also proteins, lipids, carbohydrates, and other contaminants. A commercial kit–based liquid and solid phase and automated methods have been utilized to obtain nucleic acids for hybridization techniques described in this chapter.
In liquid phase, DNA extraction detergent such as sodium dodecyl sulfate liberates DNA and proteins, which are digested by protease or salt precipitated and removed. The lipids and protein remnants can also be removed by organic extraction with phenol and chloroform. The isolated nucleic acid from free nucleus and other cellular contents is then purified by precipitation with cold ethanol. In solid-phase nucleic acid extraction method, DNA is reversibly absorbed onto coated silica onto either filters or magnetic beads at higher ionic strength solutions. The contaminants are washed away in the presence of chaotropic salts and alcohol. The DNA is then eluted by changing to low ionic strength buffer solutions and reprecipitated with cold ethanol. The purpose of these extraction methods is to isolate target nucleic acid, remove inhibitors, and concentrate nucleic acids.
There are commercial QIAamp/DNeasy kits available for extractions of genomic DNA. The buffer formulation is altered in these kits to favor genomic DNA rather than RNA binding (however, some RNA will copurify unless RNase is added during the procedure). These kits may be directed to samples that have a matrix (e.g., collagen, plant cell wall, and gram-positive bacterial cell wall), samples that have a membrane but are not embedded within a matrix (cell culture, blood, and Escherichia coli), and samples that have neither a matrix nor cell membrane (viruses, extracellular DNA in urine, or cell culture). These kits differ by the proteases included or by the addition of carrier nucleic acid to compensate for low concentrations.
The extracted nucleic acids are subjected to spectrophotometric evaluations at 260 and 280 nm for quantitation and determination of purity. The quantity of DNA and RNA is approximated by using the following estimation:
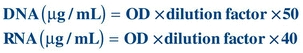
The amount of nucleic acid purity can also be assessed by calculating a ratio of optical densities at 260 nm divided by 280 nm since absorbance at 280 nm provides an estimation of protein contamination. The absorbance at 280 nm is due to amino acids tyrosine, tryptophan, and phenylalanine present in protein. The ratio of greater than 1.8 in a clinical specimen is considered to be of an acceptable purity.
For clinical assays, it is important to determine the nucleic acid degradation in an extracted specimen due to either extraction or storage. This is of concern especially for RNA preparations due to environmental presence of RNAse. The degradation is determined by ethidium bromide staining of the agarose gel electrophoresis aliquot of the extracted specimen. A DNA of good purity runs as a high molecular weight band at the origin, whereas degraded DNA will run smeared as many fragments of molecular weight bands. A high-purity RNA of eukaryotic cells demonstrates two bands of 28S and 18S.
The factors to consider for the nucleic acid extractions are usually the specimen, sample volume needed for hybridization procedures, purity, size of the nucleic acid to be isolated, ease of operation, and potential for high throughput in clinical laboratories. A number of automated workstations for nucleic acid extractions are commercially available. Many of these automated workstations can work with anticoagulated blood, which is fresh or frozen or tissue specimens, and isolate DNA or RNA in less than 1 hour. The automated isolation procedures for total RNA in some instances require the addition of genomic DNA-binding agent prior to sample loading in the automated workstation.
Hybridization Techniques
A nucleic acid probe is a short strand of DNA or RNA of a known sequence that is well characterized and complementary for the base sequence on the test target. Probes may be fragments of genomic nucleic acids, cloned DNA (or RNA), or synthetic DNA. The genomic nucleic acids are isolated from purified organisms. Some probes are molecularly cloned in a bacterial host. First, the sequence of DNA to be used as the probe must be isolated using bacterial restriction endonucleases to cut the DNA at a specific base sequence. The desired base sequence (the probe) is inserted into a plasmid vector, circular dsDNA. The vector with the insert is incorporated into a host cell, such as E. coli, where the vector replicates. The replicated desired base sequence is then isolated and purified. For short DNA segments, an oligonucleotide probe can be synthesized using an automated process; if the amino acid sequence of the protein is known, it is possible to determine the base sequence based on amino acid sequence.
The principle of hybridization is based on the reversible complementary base pairing of nucleotides. A strand of DNA (or RNA) can base-pair with a complementary oligonucleotide sequence by hydrogen bonding. The process of hybridization is controlled by the solution buffer, temperature, and the presence or absence of denaturant. High stringency conditions that include low salt concentrations, high temperatures, and the presence of denaturant such as formamide can only accept perfect complementary sequences to base-pair and hybridize. These conditions can be altered to low stringency by lowering the temperature, increasing the salt concentration, and the addition of denaturant to base-pair mismatched sequences.
In a hybridization reaction, the probe must be detected. The probe can be labeled directly with a radionuclide (such as 32P), enzyme, or biotin. 32P is detected by autoradiography when the radioactive label exposes x-ray film wherever the probe is located. If the probe is directly labeled with an enzyme, an appropriate substrate must be added to generate a colorimetric, fluorescent, or chemiluminescent product. Biotin-labeled probes can bind to avidin, which is complexed to an enzyme (e.g., alkaline phosphatase and horseradish peroxidase); the enzyme activity can then be detected. Alternatively, the biotinylated probe can be detected by a labeled avidin antibody reagent.
The probe techniques to be discussed are listed in Table 9.1. Hybridization can take place either in a solid support medium or in solution.
TABLE 9.1 Probe Techniques

Solid Support Hybridization
Dot blot and sandwich hybridization assays are the simplest types of solid support hybridization assays.3 In the dot blot assay, clinical samples are applied directly to a membrane surface. The membrane is heated to denature or separate DNA strands, and then, labeled probes are added. After careful washing to remove any unhybridized probe, the presence of the remaining probe is detected by autoradiography or enzyme assays. A positive result indicates the presence of a specific sequence of interest. This permits qualitative testing of a clinical specimen because it only indicates the presence or absence of a particular genetic sequence. It is much easier to handle multiple samples in this manner.4 However, there may be difficulty with the interpretation of weak positive reactions because there can be background interference.3
Sandwich hybridization is a modification of the dot blot procedure. It was designed to overcome some of the background problems associated with the use of unpurified samples.5 The technique uses two probes, one of which is bound to the membrane and serves to capture target sample DNA. The second probe anneals to a different site on the target DNA, and it has a label for detection. The sample nucleic acid is thus sandwiched in between the capture probe on the membrane and the signal-generating probe.5 Because of the fact that two hybridization events must take place, specificity is increased. Sandwich hybridization assays have been developed using microtiter plates instead of membranes, which has made the procedure more adaptable to automation.5
A classic method for DNA analysis, attributed to E. M. Southern, is the Southern blot.6 In this method, DNA is extracted from a sample using a phenolic reagent and then enzymatically digested using restriction endonucleases to produce DNA fragments. These fragments are then separated by agarose gel electrophoresis. The separated DNA fragments are denatured and transferred to a solid support medium—most commonly, nitrocellulose or a charged nylon membrane. The transfer occurs by the capillary action of a salt solution, transferring DNA to the membrane or using an electric current to transfer the DNA. When the DNA is on the membrane, a labeled probe is added that binds to the complementary base sequence and appears as a band. In a similar method, the Northern blot, RNA is extracted, digested, electrophoresed, blotted, and finally probed.
Restriction fragment length polymorphism (RFLP) is a technique that evaluates differences in genomic DNA sequences.7 This technique can help establish identity or nonidentity in forensic or paternity testing or to identify a gene associated with a disease. Genomic DNA is extracted from a sample (e.g., peripheral blood leukocytes) and is purified and quantitated. A restriction endonuclease, which cleaves DNA sequences at a specific site, is added. If there is a mutation or change in the DNA sequence, this may cause the length of the DNA fragment to be different from usual. Southern blotting can be used to identify the different lengths of the DNA fragments. A labeled specific probe could be used to identify a specific aberration. Polymerase chain reaction (PCR) can be used to amplify the target DNA sequence before RFLP analysis.
Solution Hybridization
Hybridization assays can also be performed in a solution phase. In this type of setting, both the target nucleic acid and the probe are free to interact in a reaction mixture, resulting in increased sensitivity compared with that of solid support hybridization. It also requires a smaller amount of sample, although the sensitivity is improved when target DNA is extracted and purified.8
For solution hybridizations, the probe must be single stranded and incapable of self-annealing.4 Several unique detection methods exist. In one of these, an S1 nuclease is added to the reaction mix. This will only digest unannealed or single-stranded DNA, leaving the hybrids intact. dsDNA can then be recovered by precipitation with trichloroacetic acid or by binding it to hydroxyapatite columns.4,9
A second and less technically difficult means of solution hybridization is the hybridization protection assay. For this assay, a chemiluminescent acridinium ester is attached to a probe. After the hybridization reaction takes place, the solution is subjected to alkaline hydrolysis, which hydrolyzes the chemiluminescent ester if the probe is not attached to the target molecule. If the probe is attached to the target DNA, the ester is protected from hydrolysis. Probes that remain bound to a specific target sequence give off light when exposed to a chemical trigger such as hydrogen peroxide at the end of the assay.
Digene Hybrid Capture 2 (Digene Corp., Gaithersburg, MD) assay for the detection of human papillomavirus (HPV) is a solution-phase hybridization assay that uses an antibody specific for DNA/RNA hybrids to “capture” and detect the hybrids that are formed during the solution hybridization of HPV DNA in the sample with an unlabeled RNA probe.
Solution-phase hybridization assays are fairly adaptable to automation, especially those using chemiluminescent labels. Assays can be performed in a few hours.4 However, low positive reactions are difficult to interpret because of the possibility of cross-reacting target molecules.10
In situ hybridization is performed on cells, tissue, or chromosomes that are fixed on a microscope slide.11 After the DNA is heat denatured, a labeled probe is added and will hybridize the target sequence after the slide is cooled. Colorimetric or fluorescent products are generally used. One strength of this method is the morphologic context in which the localization of target DNA is viewed.
DNA Sequencing
DNA sequencing is considered the “gold standard” for many molecular applications from mutation detection to genotyping, but it requires proper methodology and interpretation to prevent misinterpretation. The sequence should be analyzed on both DNA strands to provide even greater accuracy. Patient sequences are compared with known reference sequences to detect mutations. Most sequencing strategies include PCR amplification as the first step to amplify the region of interest to be sequenced. The sequencing reaction itself is based on the dideoxy chain termination reaction developed by Sanger and colleagues in 1977.12 This reaction generates fragments of newly synthesized DNA, which upon incorporation of one of four dideoxynucleotides halts synthesis of the new DNA. The fragments are of varying lengths due to incorporation of the dideoxynucleotides. These nucleotides lack the 3′- and 2′-hydroxyl (OH) group on the pentose ring, and because DNA chain elongation requires the addition of deoxynucleotides to the 3′-OH group, incorporation of the dideoxynucleotide terminates chain length. Varying lengths of DNA are synthesized, all with a dideoxynucleotide at the end. The reaction mixture is then run on a gel, separating the various DNA fragments by size, and the sequence is read directly from the gel. The most commonly used form of this sequencing method in the clinical laboratory uses “cycle sequencing,” which is similar to a PCR in that the steps involved include denaturation, annealing of a primer, chain extension, and termination by varying the temperature of the reaction. The newly generated fragments are tagged with a fluorescent dye and separated, based upon size, by denaturing gel or capillary electrophoresis and detected by fluorescence detectors as the fragments pass through the detector. Using this automated method with capillary electrophoresis, about 600 base pairs can be sequenced in a 2½-hour period (Fig. 9.2). DNA sequencing is most commonly used to detect mutations. For example, in infectious disease, testing such as sequencing of the human immunodeficiency virus (HIV) for drug resistance and of the hepatitis C virus (HCV) is used to establish appropriate therapy and treatment decisions.
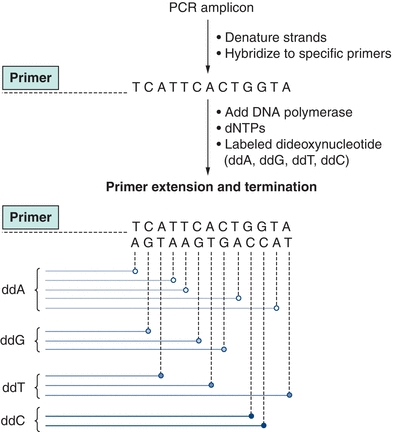
FIGURE 9.2 Sanger dideoxy DNA sequencing. The region of interest is first amplified using polymerase chain reaction. Sequence-specific primers then hybridize with the denatured amplicons, followed by extension of the new strand by DNA polymerase. At various points in the DNA extension, a dideoxy base (ddA, ddG, ddT, or ddC) is incorporated, which stops further extension of DNA. This results in a mixture of newly synthesized products of various lengths. Each dideoxy terminator base is labeled with a specific fluorescent tag, which can be detected with a fluorescent detector. The fragments are separated based upon their size, and fluorescence is read on each strand, resulting in the sequence of the DNA.
Pyrosequencing1,2 is a relatively new technique for a short to moderate sequence analysis that is based on the release of pyrophosphate during DNA synthesis as each dNTP (deoxyribonucleotide triphosphate) is incorporated with elongation of DNA. In this method, after primer annealing to template strand and with subsequent addition of the complementary dNTPs toward the 3′-end by DNA polymerase, PPi (pyrophosphate) is released as the nucleotide forms a phosphodiester bond with the primer. Sulfurylase and its substrate adenosine 5′-phosphosulfate then convert PPi to ATP, which subsequently transforms luciferin to oxyluciferin by luciferase. This conversion of oxyluciferin generates a luminescent signal proportional to the amount of ATP. The light signal is detected by a charge-coupled device and seen as a peak in a pyrogram. The number of nucleotides incorporated is proportional to the height of each peak observed. Apyrase degrades unincorporated nucleotides and ATP. When degradation is completed, another nucleotide is added. As the complementary DNA (cDNA) is synthesized, the nucleotide sequence is determined from the signal peaks in real time by built-in computer software. The technique does not require gel electrophoretic separation of ddNTPs (dideoxyribonucleoside triphosphate), DNA fragments, and radioactive or fluorescent labels. The pyrosequencing technique is useful for the detection of SNP and has been applied in the diagnosis of infectious diseases and human leukocyte antigen typing.
Next-Generation Sequencing
Sanger developed biochemical sequencing methods are now replaced by a new technology where fragmented DNA is sequenced in massively parallel reactions with the use of oligonucleotide adapters. This next-generation sequencing (NGS) can generate a whole genome or exome (coding region) sequence. There are a variety of commercial instruments available for NGS that utilizes sequencing-by- synthesis chemistries. These instruments utilize an extremely sensitive CCD camera to detect light emitted from the reaction.
Roche developed FLX+ 454 Genome Sequencer uses PCR amplification cycle products on an etched glass fiber with beads. The generation of a pyrophosphate group emits light after the introduction of a known unlabeled nucleotide one at a time. The light signal identifies the incorporation of the base in an extended DNA strand. HiSeq 2000 System developed by Illumina utilizes reversible specific detectable fluorescent dye–based nucleotide terminator. The amplification process provides enhanced light signal with every PCR cycle. This method has a very high throughput that can read in the order of gigabases in a short period of time. MiSeq also developed by Illumina is a smaller version of HiSeq, but with similar throughput. In contrast, Personal Genome Machine by Ion Torrent builds on an amplified cDNA library. A new DNA fragment in this technology is synthesized from a template provided by the library with a detection that relies on the changes in pH due to the release of a H+ ion during nucleotide incorporation.
DNA Chip Technology
Previously, genetic studies examined individual gene expression by Northern blotting or polymorphisms by gel-based restriction digests and sequence analysis, but with advances in microchips and bead-based array technologies, high-throughput analysis of genetic variation is now possible. Biochips, also called microarrays, are very small devices used to examine DNA, RNA, and other substances. These chips allow thousands of biological reactions to be performed at once.13 Typically, a biochip consists of a small rectangular solid surface that is made of glass or silicon with short DNA or RNA probes anchored to the surface.14 The number of probes on a biochip surface can vary from 10 to 20 up to hundreds of thousands.13,14 Usually, the nucleic acid in the sample is amplified before analysis. After amplification, the sample, labeled with a fluorescent tag, is loaded onto the chip. Hybridization occurs on the surface of the chip, allowing thousands of hybridization reactions to occur at one time. Unbound strands of the target sample are then washed away. The fluorescent tagged hybridized samples are detected using a fluorescent detector.13 The intensity of the fluorescent signal at a particular location is proportional to the sequence homology at a particular locus. Complete sequence matches result in bright fluorescence, while single-base mismatches result in a dimmer signal, indicative of a point mutation. Because most SNPs are silent and have no apparent functional consequence, the challenge is to identify the set of SNPs that are directly related to or cause disease. Detection of point mutations can be used for classification of leukemias, molecular staging of tumors, and characterization of microbial agents.15 One prime example is the determination of genes associated with drug resistance in HIV testing. Identification of such genes guides the physician in selecting a proper drug regimen for a particular patient.
Microarrays
In this technology, thousands of known unlabeled 5 to 50 bp DNA sequence probes are immobilized on a solid support to which clinical specimens are incubated for hybridization. The solid support can be a silicon chip or a nylon membrane that holds different probes at a specific location allowing multitudes of hybridization reactions. Microarrays (1) are clinically used to detect genetic polymorphism and disease-specific spectrum of mutations simultaneously (2) can perform comparative genomic hybridization to evaluate genomic gains and losses and determine molecular karyotype.
Currently, Affymetrix, Inc., has developed GeneChips, proprietary, high-density microarrays that contain 10,000 to 400,000 different short DNA probes on a 1.2-cm × 1.2-cm glass wafer. GeneChips are available for HIV-1 genotyping,15 human p53 tumor suppressor gene mutation analysis, and human cytochrome p450 gene mutation analysis, with other GeneChips currently under development.
Target Amplification
Target amplification systems are in vitro methods for the enzymatic replication of a target molecule to levels at which it can be readily detected.8 This allows the target sequence to be identified and further characterized. There are numerous different types of target amplification. Examples include PCR, transcription-mediated amplification (TMA), SDA, and nucleic acid sequence–based amplification (NASBA). Of these, PCR (Table 9.1) is by far the best known and most widely used technique in clinical laboratories.16 However, the other non-PCR methods have become more popular in recent years.
The PCR, developed by K. B. Mullis of Cetus Company,17,18 is an amplified hybridization technique that enzymatically synthesizes millions of identical copies of the target DNA to increase the analytic sensitivity. The test reaction mixture (Table 9.2) includes the test DNA sample (lysed cells or tissue enzymatically digested with RNase and proteinase and then extracted), oligonucleotide primers (Table 9.3), thermostable DNA polymerase (e.g., Taq polymerase, from Thermus aquaticus), and nucleotide triphosphates (ATP, GTP, CTP, and TTP) in a buffer. The process, shown in Figure 9.3, begins by heating the target DNA to denature it and then separating the strands. Two oligonucleotide primers (probes) that recognize the edges of the target DNA are added and anneal to the target DNA. Thermostable DNA polymerase and nucleotide triphosphates extend the primer. The process of heat denaturation, cooling to allow the primers to anneal and heating again to extend the primers, is repeated manyfold (15 to 30 times or more). PCR is an exponential amplification reaction in which after n cycles, there is (1 + x)n times as much target as was present initially, where x is the mean efficiency of the reaction for each cycle. Theoretically, as few as 20 cycles would yield approximately 1 million times the amount of target DNA initially present. However, in reality, the theoretical maxima are never reached and more cycles are necessary to achieve such levels of amplification. The amplified target DNA sequences, known as amplicons, can be analyzed by gel electrophoresis, Southern blot, sequencing, or directly labeled probes.
TABLE 9.2 Real-Time Polymerase Chain Reaction Components

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


