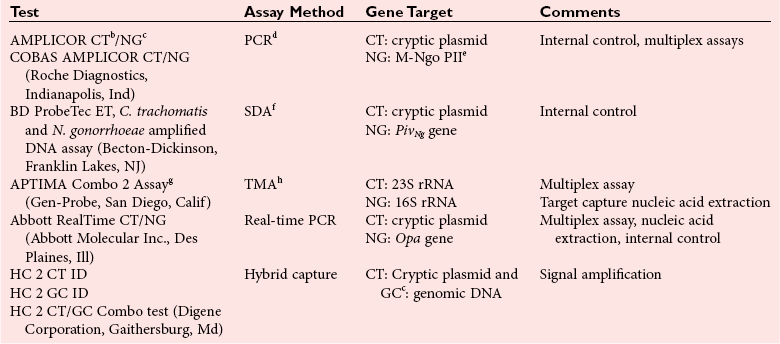
Chapter 42 Interpretation of the results of molecular assays for infectious diseases requires an understanding of the biology of the target organism, the pathogenesis of related infectious disease(s), and the advantages and limitations of the technology used. An advantage of molecular methods is their exquisite sensitivity for the detection of pathogen nucleic acids whether from viable or nonviable organisms. The nucleic acids of the organism may persist for varying lengths of time after adequate treatment of an infection. Therefore, one of the challenges can be interpreting the significance of a positive molecular test result. For example, Chlamydia trachomatis DNA can be detected in the urine of patients for as long as 3 weeks after initiation of appropriate therapy.48 Similarly, herpes simplex virus (HSV) DNA can be detected in the cerebrospinal fluid (CSF) of patients with encephalitis for 2 weeks or longer after initiation of acyclovir therapy.84 Thus, in this case and most others, monitoring response to therapy with qualitative assays has limited clinical utility and is best done using quantitative methods. Detection of the nucleic acid of a pathogen does not ensure that the organism is the cause of the disease. The organism may be part of the normal flora, colonizing a specific area or causing infection but not disease. For example, interpretation of molecular tests for the detection and quantification of herpesviruses after primary infection is complicated by the fact that these viruses establish a life-long latent infection. Early studies evaluating the clinical utility of cytomegalovirus (CMV) DNA assays used very sensitive qualitative methods and peripheral blood mononuclear cells as the specimen of choice. As a result, CMV DNA was detected in immunocompromised patients with disease, as well as in those without CMV disease. Studies are needed to establish viral load values that distinguish between latent infection, asymptomatic disease, and active disease. Factors that need to be considered when a positive result is interpreted include assay specificity and contamination. Specificity of molecular infectious disease assays is related to the primers and probes used during amplification and detection/quantification steps. If primers allow amplification of nucleic acids from other pathogens normally present in a patient specimen, false-positive results are possible. Although uncommon, problems with primer specificity have been reported; primers designed to amplify the 5′ untranslated region of the enterovirus genome have also been shown to amplify rhinovirus RNA.129 This will not be an issue if the assay is used only for cerebrospinal fluid, but it will likely cause false-positive results if respiratory specimens are tested. Detection of CT is a challenging and important public health issue. CT is a major cause of genital infections, with an estimated 1 million cases occurring annually among sexually active adolescents and young adults in the United States.53 More than half of the infections are asymptomatic.144 Even when symptomatic, the diagnosis can be missed as the manifestations are protean. In males, CT infection may present as urethritis, epididymitis, prostatitis, or proctitis17,105 and as cervicitis, endometritis, and urethritis in women, with 10 to 40% of infections in women progressing to pelvic inflammatory disease (PID) if untreated.122,145 Related complications include chronic pelvic pain, ectopic pregnancy, and infertility. In the United States, CT infection is a likely cause of most secondary infertility in females. In pregnant women, there is the additional risk of transmitting the infection to the newborn during labor and delivery, leading to pneumonia or conjunctivitis in the newborn. Traditional methods for the diagnosis of CT infection include cell culture, antigen detection by immunofluorescence-based techniques, enzyme immunoassay, and, more recently, nonamplified nucleic acid detection. These traditional methods have been replaced in many laboratories by amplified nucleic acid tests, which provide greater sensitivity in detecting CT from genital specimens. For GC, which was traditionally diagnosed based on culture methods that relied on selective culture media, nucleic acid testing does not offer significant improvement in sensitivity compared with culture when culture is performed under ideal conditions. GC is a fastidious organism, however, and is highly susceptible to extreme temperatures and desiccation, which can lead to decreased sensitivity of detection by culture, particularly when specimen transport is required prior to culturing.76 Nucleic acid testing for GC offers a sensitive and reliable alternative to culture. Tests for the detection of CT and GC from clinical specimens (Table 42-1) use a variety of specimens, including cervical and vaginal swabs, urethral swabs, and urine from both asymptomatic and symptomatic individuals. Not all assays are cleared by the U.S. Food and Drug Administration (FDA) for use in the United States for all conditions, and the current assays are not FDA-cleared for oral, rectal, respiratory, or conjunctival specimens. Performance characteristics vary among assays (details are available in the package inserts), but some general comments can be made. The diagnostic sensitivity of the tests varies according to the specimen type and whether the patient is asymptomatic or symptomatic. Interpretation of the results of nucleic acid testing for CT can be challenging because many studies have shown these assays to be more diagnostically sensitive than culture, which was previously used as the gold standard for clinical trials. For males, the diagnostic sensitivity of testing urine specimens is nearly equivalent to that of testing urethral swabs.16,29,36,72,155 A limited volume (20 to 50 mL) of first-passed urine is preferred because larger volumes will lead to a decreased concentration of the organism in the sample and thus reduced diagnostic sensitivity. With proper specimen collection, male urethral swabs and urine specimens have a sensitivity of nearly 100% for the detection of GC or CT infection. For women, cervical swab specimens provide the highest sensitivity for the detection of GC and CT infection, with many studies showing a sensitivity of 90 to 95%.29,36,86,115 Urine specimens can be used, but they generally result in a lower diagnostic sensitivity than cervical swabs (75 to 85%).29,36,86,115 An alternative to urine testing in women is the use of self-collected vaginal swabs, which have been shown in some studies to have a diagnostic sensitivity that is equal to that obtained with cervical swabs; several of the tests have been cleared for use with vaginal swabs.65,134 TABLE 42-1 Summary of FDA-Cleared Testing for Chlamydia trachomatis (CT) and Neisseria gonorrhoeae (NG)a aFor FDA listings of these and any newer tests, see http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfRL/listing.cfm. Use test codes LSK and LSL for C. trachomatis and N. gonorrhoeae, respectively. eCytosine DNA methyltransferase. gConfirmatory assays for CT and NG are also available. For several of the GC assays, reduced specificity is due to cross-hybridization of primers with nongonococcal Neisseria species.56,104 The ProbeTec test (Becton-Dickinson, Franklin Lakes, NJ) has been reported to produce false-positive results with N. flavescens, N. lactamica, N. subflava, and N. cinerea, and the AMPLICOR assay (Roche Diagnostics, Indianapolis, Ind) can produce false-positive results with N. flavescens, N. lactamica, and N. sicca. There is concern about generating false-positive results with pharyngeal specimens that may contain these nongonococcal species of Neisseria. However, N. cinerea, N. lactamica, N. subflava, and N. sicca have also been isolated from genital mucosa, so it is possible to generate false-positive results from genital specimens. Nucleic acid testing for the detection of CT or GC should not be used as a test of cure. Because DNA can persist in urine samples for up to 3 weeks after completion of therapy,48 test of cure using nucleic acid testing is discouraged. If this must be done, then testing should be delayed for at least 3 weeks after therapy is begun to allow time for clearance of the DNA of the pathogen. False-negative results from inhibition of amplification are a consideration for both GC and CT testing and have been reported for both cervical swabs and urine specimens.30,89,127 Inhibition rates may vary considerably depending on the amplification method used and are related, in part, to the method used for nucleic acid extraction.89 For tests that use a crude lysate in testing (such as the AMPLICOR and ProbeTec tests), inhibition rates tend to be higher than those seen with the APTIMA Combo test (Gen-Probe Inc., San Diego, Calif), which uses a target capture method to purify nucleic acid. For assays that test a crude lysate, it is useful to amplify another nucleic acid sequence as an internal control (or “amplification control”) to assess for inhibition of amplification. Results are reported as negative for GC or CT only when amplification of the internal control is documented. A variant strain of CT has been identified in Sweden with a 377-base-pair deletion in the cryptic plasmid, which is the target for several of the CT tests. This deletion leads to false-negative results with some but not all of the tests that target the cryptic plasmid. Tests that target other regions of the organism are not affected. The ability of a test to detect this variant is important when a test is chosen, particularly if this variant is commonly found in the geographic area where the test will be used.91,124 Performing CT and GC testing for liquid cytology specimens is a matter of interest because a single specimen can be used for cervical cytology (PAP smear) and for CT and GC testing.8 The latter two tests would be performed on the liquid specimen that remained after completion of the PAP and human papillomavirus testing. However, several drawbacks to this approach must be considered. The instruments used to prepare liquid PAP smears were not designed to control for cross-contamination during processing, and this may lead to false-positive results. CT and GC testing would not be performed until after the PAP smear and human papillomavirus testing were completed, which could delay diagnosis and treatment of CT or GC infection. Moreover, the remaining specimen may be inadequate to complete CT and GC testing, thus requiring the patient to make a return visit for collection of an additional sample. Removing an aliquot for CT and GC testing before PAP testing is performed may be helpful in overcoming some of these issues, provided adequate volume of sample remains for PAP testing. This approach does not completely remove the risk of cross-contamination, so specimens must be handled in a manner consistent with procedures used in molecular laboratories, such as uncapping and aliquoting one specimen at a time. In January of 2009, an expert panel was convened by the Association of Public Health Laboratories in cooperation with the Centers for Disease Control and Prevention to update recommendations on laboratory diagnostic testing for CT and GC (document available at www.aphl.org; Laboratory Diagnostic Testing for Chlamydia trachomatis and Neisseria gonorrhoeae). Several of the new and updated recommendations are as follows: 1. Urine is the preferred specimen in males. Vaginal swabs are the preferred specimen for screening females, because the sensitivity of tests with vaginal specimens is equal to or superior to that with cervical swabs. 2. Nucleic acid testing (NAT) is recommended for the detection of rectal and oropharyngeal infections caused by CT and GC. Because currently available CT and GC tests are not cleared for this specimen type, laboratories must perform a validation study to establish the performance characteristics of the test. A NAT test with minimal cross-reactivity with commensal Neisseria species should be used. 3. Routine repeat testing of NAT-positive screening tests is not recommended. 4. NAT testing is superior to culture for the detection of CT in cases of adult rape or abuse. For GC, positive results of tests that have significant cross-reactivity with nongonococcal Neisseria species should be retested with a different NAT. 5. In cases of pediatric sexual abuse, NAT for CT has been shown to be superior to culture. For GC, positive results of tests that have significant cross-reactivity with nongonococcal Neisseria species should be retested with a different NAT. Human papillomaviruses (HPVs) are small, double-stranded DNA viruses that infect squamous epithelium, subverting normal cell growth and potentially leading to squamous cell carcinoma.98 Anogenital HPV infections are common in both men and women. It is estimated that more than 24 million men and women in the United States are currently infected with HPV. HPV is a sexually transmitted infection; it is most common among sexually active young women ages 15 through 25 years. In one study, cervicovaginal HPV was found in up to 43% of sexually active college women during a 3-year period.63 Infections, however, are usually transient, and progression to cancer requires persistence of viral infection over several years. The types of HPV that are spread through sexual contact are classified as low risk or high risk for progression to malignancy, and there are multiple types. Infections with low-risk HPV such as types 6 and 11 can lead to benign genital warts or condyloma acuminata and have a low likelihood of progressing to malignancy. In contrast, high-risk types such as types 16, 18, and 45 are associated with development of squamous cell carcinoma of the anogenital region and oropharynx. The cervix is particularly affected, and worldwide cervical squamous cell carcinoma continues to cause significant morbidity and mortality (5% of cancer deaths). Primarily as a result of the ALTS trial (ASCUS and LSIL Triage Study), performed in the late 1990s, HPV testing now plays an important role in assessing which women with ASCUS are at highest risk of developing cervical cancer. The 2006 Guidelines recommend that women 20 years or older in whom ASCUS is found on Pap testing should be tested for the presence of high-risk HPV.167 Those women testing positive for high-risk HPV DNA should undergo further clinical and pathologic examination (colposcopy), and those testing negative for HPV DNA can be followed according to routine practice. The Guidelines also recommended that women 30 years or older with a normal Pap and a negative HPV test result could have less-frequent examinations (every 3 years), and those with an HPV-positive test result could have (1) repeat Pap or HPV testing in a year, or (2) HPV genotyping performed; if found to be positive for type 16 or 18, they should be referred for immediate colposcopy. Three tests for the detection of HPV DNA and one for genotype identification of HPV 16 and 18 have been cleared by the FDA for use in the United States: Hybrid Capture 2 (Qiagen Inc., Valencia, Calif), Cervista HPV HR (Hologic Inc., Danbury, Conn), cobas® HPV Test (Roche Diagnostics) and Cervista HPV-16 and -18 Genotyping test. All four tests have been cleared by the FDA for use with ThinPrep PreservCyt liquid-based cytology media (Hologic Inc.), but not with the other commonly used SurePath media (Becton-Dickinson). The Hybrid Capture 2 (HC2) test relies on hybridization of an RNA probe to the HPV DNA, followed by use of an antibody for capture of the duplex (RNA-DNA) hybrids, and then detection with chemiluminescent signal amplification. The test uses a pool of RNA probes spanning the entire genome that are specific for 13 high-risk HPV types (16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59, and 68). The specific type is not identified. The test uses a 96-well microtiter plate format and can be performed manually or with the semiautomated Rapid Capture system (Qiagen) for reagent and plate handling. It is also cleared for use on Digene specimen transport media (STM). The HC2 test has been used in several large studies and reproducibly demonstrates high sensitivity of 93 to 96%, but false-positive results occur as a result of cross-reaction with low-risk HPV types.23 Human immunodeficiency virus type 1 (HIV-1), the causative agent of the acquired immunodeficiency syndrome (AIDS), is an RNA virus belonging to the genus Lentivirus of the family Retroviridae. Replication of the virus is complex and involves reverse transcription of the RNA genome into a double-stranded DNA molecule or provirus, which is integrated into the host genome. HIV-1 enters the cell using CD4 as a receptor and CXCR4 or CCR5 as a coreceptor. In general, CCR5 coreceptors are found on macrophages, and CXCR4 coreceptors are found on T cells. Determining the cellular tropism of the virus has become important, now that a new antiretroviral drug targets the CCR5 coreceptor. The HIV-1 reverse transcriptase enzyme does not have proofreading capabilities, leading to the marked genetic diversity of HIV-1. Several distinct genetic subtypes or clades have been identified and are categorized into three groups: major (M), outlier (O), and N (nonmajor and nonoutlier). The major group is divided into nine clades (A, B, C, D, F, G, H, J, and K) and circulating recombinant forms (CRFs), which are determined on the basis of sequence diversity within the HIV-1 gag and env genes.42 Group M virus is found worldwide, with clade B predominating in Europe and North America, clade C in Africa and India, and clade E in much of Southeast Asia. Complex replication cycles and genetic diversity are two factors that influence the design and interpretation of HIV-1 molecular assays. Viral load testing became the standard of care around 1996, followed by resistance testing. The clinical utility of viral load testing (usually using blood plasma) has been well established. Testing is used (1) to determine when to initiate antiretroviral therapy, (2) to monitor response to therapy, and (3) to predict time to progression to AIDS. Higher viral loads are associated with more rapid progression to AIDS and death.99,100,111 Viral load testing is used routinely in decisions regarding when to initiate antiretroviral therapy and in monitoring response to therapy. Current treatment guidelines [U.S. Department of Health and Human Services (DHHS) Panel on Antiretroviral Guidelines, http://AIDSinfo.nih.gov] recommend initiating therapy for individuals on the basis of several factors, including concentrations of CD4-positive cells in blood (CD4 cell counts), viral loads, and symptoms. The current standard for treating HIV-1–infected individuals is to use combinations of highly active antiretroviral drugs. Multiple classes of drugs are used, including nucleoside reverse transcriptase inhibitors (NRTIs), non-nucleoside reverse transcriptase inhibitors (NNRTIs), protease inhibitors (PIs), fusion inhibitors, integrase inhibitors, and CCR5 entry inhibitors. Current guidelines (DHHS Panel on Antiretroviral Guidelines, http://AIDSinfo.nih.gov) recommend an initial regimen of two NRTIs and either an NNRTI or a PI. This combination therapy is often referred to as highly active antiretroviral therapy, or HAART. Initial use of these effective drug combinations in individuals who have not been treated with them before (“naive” individuals) is expected to decrease viral loads by at least 100-fold, or 2 log10 copies/mL. The goal of therapy is to achieve viral loads below the limit of detection of currently available assays (50 copies/mL), although this is not always possible in all individuals, particularly in those with very high pretreatment viral load values, or in those who have failed prior therapeutic regimens. Guidelines for the use of HIV-1 RNA viral load values in clinical practice have been published131 and are frequently updated (http://www.aidsinfo.nih.gov/, http://www.iasusa.org). In general, a plasma HIV-1 viral load should be measured before therapy is begun (baseline), and then again at 2 to 8 weeks after initiation of therapy to determine the response to therapy. Testing is then repeated at 3- to 4-month intervals to evaluate continued effectiveness of the regimen. Any increase in viral load should be confirmed with repeat testing because a variety of other illnesses can transiently increase viral load. When a significant increase in viral load has been documented, HIV-1 resistance testing should be considered (see later). The use of HIV-1 viral load testing for diagnosing acute HIV-1 infection in adults is more controversial. Currently available viral load assays are approved by the FDA for use only in patients known to be infected with HIV-1, but they have clear utility in the diagnosis of acute infection, which is defined as the period after exposure to the virus but before seroconversion, that is, while the enzyme-linked immunosorbent assay (ELISA) and Western blot assays for antibodies to HIV-1 are negative or indeterminate. In this “window period,” additional testing is required and viral load assays are often used. Individuals with acute infection are often symptomatic with a mononucleosis-type syndrome, which may include fever, fatigue, rash, lymphadenopathy, and oral ulcers.77 During this acute infection, the plasma concentration of viral RNA is very high, usually 105 to 107 copies/mL, and viral load measurements are a useful diagnostic tool. Acute HIV-1 infection should be suspected in an individual presenting with appropriate symptoms and risk factors. In these individuals, testing for acute HIV-1 infection would include an ELISA and a viral load assay. Care must be taken to correctly interpret these test results, because individuals with acute HIV-1 infection would be expected to have a negative or indeterminate ELISA and/or Western blot, and a very high viral load (>100,000 copies/mL). The concern with using viral load testing to diagnose acute HIV infection is that false-positive results have been reported.37 In one study, false-positive results (usually lower than 2000 copies/mL) were found when the VERSANT bDNA test (Siemens Healthcare Diagnostics, Deerfield, Ill) was used.37 Before acute HIV infection is diagnosed, individuals must be educated regarding the limitations of these tests and must give informed consent prior to testing. To minimize the likelihood of reporting a false-positive result, repeat testing should be done on all specimens with a detectable viral load, and an HIV-1/-2 ELISA should be obtained at the time of viral load testing. It is critical to remember that patients with acute retroviral syndrome should have very high concentrations of HIV-1 RNA. HIV-1 viral load assays that are currently approved by the FDA are listed in Table 42-2. (FDA-approved devices have been found by the FDA to be safe and effective for a defined clinical use. Many of the other tests described in this chapter are FDA-cleared devices; they have been deemed substantially equivalent to an existing “predicate” device.) In addition to conventional molecular tests [AMPLICOR MONITOR, and VERSANT], two real-time HIV-1 viral load tests have been approved by the FDA. The lower limit of quantification differs among the assays, although most tests reach levels of 40 to 50 copies/mL. The reportable concentration range of each of the conventional AMPLICOR assays is limited, so both an ultrasensitive and a standard version of the test are needed to cover the clinically important range of viral load values. Viral load assays must be able to accurately quantify the various viral subtypes. In the United States and Europe, subtype B predominates, although infections with non-B subtypes are becoming more common and are certainly an important cause of HIV-1 infection globally. The VERSANT bDNA assay will accurately quantify HIV-1 subtypes A through G,71 and the AMPLICOR reverse transcriptase polymerase chain reaction (RT-PCR), version 1.5 (Roche Diagnostics), will perform accurate quantification of subtypes A through H.71,109,113,114 The real-time RT-PCR tests have been designed to detect genetically diverse types and subtypes of HIV-1. The COBAS TaqMan test (Roche Diagnostics) quantifies all subtypes of group M and group N viruses and many CRFs.137 Version 2 of the COBAS TaqMan test is now available and has a lower limit of quantification of 20 copies/mL and accurately quantifies Group O virus and a broader range of CRFs compared to the version 1 test. The RealTime TaqMan test quantifies all group M and N viruses, CRFs, and group O virus.54,149 Real-time viral load tests have several other important advantages compared with conventional viral load tests, including broader linear range, greater automation, and decreased risk of carryover contamination. Viral load values obtained with the different assays may not always agree, so it is recommended that one assay is chosen when patients are monitored over time. TABLE 42-2 Quantitative HIV (Viral Load) Assays Approved by the FDA* *For FDA listings of these and any newer tests, visit http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfRL/listing.cfm and use test code MTL. †Reverse transcriptase polymerase chain reaction. Analytical characteristics of viral load assays have been examined in several studies.14 The intra-assay imprecision (SD) of the assays is approximately 0.12 to 0.2 log10, with the VERSANT bDNA assay showing the best precision. The biological variation (as SD) of the virus in patients not receiving therapy is approximately 0.3 log10 copies/mL.88,131 For the AMPLICOR (version 1.0) assay, the total variation was approximately 0.26 log10, including intra-assay, interassay, and biological variation, with biological variation accounting for 56 to 80% of the total variation.15 Changes in viral load must exceed 0.5 log10 (a fivefold change) or more to suggest a change in viral replication rather than a change in viral load attributable to analytical and day-to-day biological variation. Reporting viral load results as log10 copies/mL is recommended33 and will assist in preventing clinicians from overinterpreting small changes in viral load. This is particularly important for values near the limit of quantification, where assay variability is the greatest. A variety of acute and opportunistic infections and vaccinations can transiently increase HIV-1 RNA in plasma,39,111,147 so it is recommended not to measure viral load for monitoring of individuals who are acutely ill and those who have been recently vaccinated. Viral load testing is routinely performed on plasma specimens, and ethylenediaminetetraacetic acid (EDTA) is the anticoagulant of choice. Acid-citrate-dextrose is also an acceptable anticoagulant, but blood anticoagulated in heparin is unacceptable for most tests. Viral load testing can be done on specimens other than plasma, including serum, dried blood (or plasma) spots on filter paper, CSF, and genital secretions. The assays have not been approved by the FDA for all of these specimen types, and testing of these specimens is usually reserved for research studies. It is critical to handle clinical specimens properly to minimize the risk of RNA degradation during specimen collection and transport. Plasma should be separated within 6 hours of collection and ideally stored at −20 °C, although plasma viral RNA is stable at 4 °C for several days. For laboratories performing testing from specimens collected at remote sites, sample handling can require careful attention. Special blood collection containers, or tubes, are available that contain a gel that provides a physical barrier between the plasma and cells after centrifugation. The tubes can be shipped without the need to transfer the plasma into a separate tube.64 Vacutainer brand plasma preparation tubes (PPTs) (Becton-Dickinson), similar to plasma separation tubes (PSTs), are an example of such a container. Tubes should not be frozen prior to pouring off of the plasma, as this may lead to a false increase in results of viral load assays.47,52 Both qualitative RNA and proviral DNA tests are useful for the diagnosis of HIV-1 infection in newborns (Guidelines for the Use of Antiretroviral Agents in Pediatric HIV Infection, http://AIDSinfo.nih.gov). Because maternal immunoglobulin (Ig)G crosses the placenta, an uninfected child of an HIV-positive mother may be seropositive into the second year of life. The diagnosis or exclusion of HIV-1 infection requires use of testing at several different time points, usually (1) shortly after birth (14 to 21 days), (2) at 1 to 2 months, and (3) at 4 to 6 months after birth. The diagnosis of HIV-1 infection requires two positive RNA or DNA tests performed on separate blood samples regardless of age. RNA and DNA tests have different strengths and weaknesses. HIV-1 DNA tests may be less sensitive for the detection of non-B subtype infections than are HIV-1 RNA tests, particularly the FDA-approved APTIMA HIV-1 RNA Qualitative Assay (Gen-Probe), which is the first qualitative NAT approved by the FDA for the diagnosis of acute HIV-1 infection. Proviral DNA tests are also useful for the diagnosis of neonatal infection because they remain positive when the infant is receiving antiretroviral therapy. Currently, no proviral DNA tests have been approved by the FDA; the AMPLICOR HIV-1 DNA PCR assay (Roche Diagnostics) is available as a research-use-only (RUO) test. A variety of studies have evaluated the clinical utility of antiviral resistance testing in HIV-1–infected individuals. Several early prospective randomized clinical trials of genotypic resistance testing were conducted with persons who had failed therapy with multidrug regimens including PIs and NRTIs. In both the VIRADAPT32,40 and GART6 studies, selection of the salvage regimen was determined by using genotypic resistance testing (genotype arm) or by considering which antiretroviral drugs had been used in prior treatment regimens (control arm). Response rates in the genotype arms were higher than in the control arm. For example, in the VIRADAPT study, patients in the genotype arm had a greater decrease in viral load 6 months after salvage therapy was initiated, and more of them (32% vs. 14 %) had plasma viral loads <200 copies/mL.32,40 The HAVANA trial helped establish the utility of expert advice in interpreting genotypic resistance data by comparing genotype resistance testing, expert advice, or both with the standard of care for selecting a regimen in patients failing therapy.154 Although either genotyping or expert advice improved response compared with the control group, the best response was seen in the group receiving both genotyping and expert advice. The VIRA3001 study, a prospective randomized trial that compared standard of care versus phenotypic resistance testing in patients who failed a PI-containing regimen, found a better virologic outcome for patients in the phenotypic arm.34 Although some trials of resistance testing have not shown improved clinical outcomes compared with standard of care,59,101,103 the results of randomized trials favor use of resistance testing. Guidelines for the appropriate use of HIV-1 resistance testing in adults have been established and are regularly updated (DHHS Panel on Antiretroviral Guidelines, http://AIDSinfo.nih.gov). Resistance testing is recommended in the following situations: (1) when changing antiretroviral regimens, (2) before initiating therapy in patients who have never received therapy, (3) during management of patients who have not obtained an optimal decrease in viral load values, and (4) before initiating therapy in all pregnant women. Interpretation of genotypic resistance testing is complex. Interpretation of resistance mutations uses “rules-based” software that takes into account cross-resistance and interactions of mutations. The commercially available systems generate a summary report that lists the various mutations that have been identified in the reverse transcriptase and protease genes, and each drug is reported as resistant, possibly resistant, no evidence of resistance, or insufficient evidence. A comprehensive discussion of the specific mutations associated with each antiretroviral drug and the interactions of mutations is beyond the scope of this chapter, but is available from a variety of sources (http://www.iasusa.org, http://hivdb.stanford.edu/). Phenotypic resistance assays measure viral replication in the presence of antiretroviral drugs. Results of phenotypic assays are typically reported as the inhibitory concentration of a drug that reduces in vitro HIV-1 replication by 50% (IC50). The IC50 is usually reported as the fold change in IC50 relative to a wild-type strain. Initially, phenotypic assays required the isolation of infectious HIV-1 from a blood specimen. Newer phenotypic assays use high-throughput automated assays based on recombinant DNA technology. For these assays, HIV-1 RNA is amplified from a plasma specimen, eliminating the need for a viral isolate. This testing is not performed in clinical laboratories, and the technology is available from only two commercial laboratories. For the PhenoSense assay (Monogram Biosciences, South San Francisco, Calif), the protease and reverse transcriptase genes are amplified using RT-PCR and are inserted into a modified HIV-1 vector that has a luciferase reporter gene in place of the viral envelope gene.117 Drug resistance is assessed by quantification of luciferase expression in the presence of various concentrations of antiretroviral drugs. The reproducibility of the assay is such that increases in IC50 greater than 2.5-fold can be reliably detected in the assay. The other assay (Antivirogram, VIRCO Lab Inc., Bridgewater, NJ) combines patient and HIV-1 vector sequences using in vitro recombination.34 Viral replication is measured using a reporter gene system. Based on replicate studies performed by the company, reduced susceptibility is defined as a greater than fourfold increase in IC50 compared with wild-type virus. This technical cutoff often differs from the cutoff that is associated with clinical resistance to a drug, which is referred to as the clinical cutoff. The change in IC50 associated with clinical failure may differ for each drug tested. For example, with the protease inhibitor lopinavir, the IC50 that correlates with clinical resistance may be in the range of a 10-fold or greater increase in IC50,156 compared with twofold for the NRTI didanosine (ddI). It is likely that IC50 cutoffs will continue to be modified as more clinical outcomes data become available. Results of phenotypic assays include not only the change in IC50 value, but an interpretation of whether there is an increase or decrease in susceptibility compared with wild-type virus. Phenotypic tests are available to measure susceptibility to NRTIs, NNRTIs, PIs, fusion inhibitors, and integrase inhibitors. Both phenotypic and genotypic assays are used in clinical care. Some clinicians prefer phenotypic testing because it is a direct measure of viral susceptibility; others prefer genotypic testing because the development of a mutation may precede phenotypic expression of resistance.62 Other advantages of genotypic testing include relatively rapid turnaround time (a few days), easier availability, and lower cost compared with phenotypic testing. Providers often use genotypic testing routinely and rely on phenotypic testing for patients who have failed multiple regimens and have very complex genotypic results. If both assays are used, it is important to remember that the results of the two assays may not agree, as the presence of a resistance mutation does not ensure its expression in a phenotypic assay. Maraviroc, the new CCR5 inhibitor, is effective only against HIV-1 that uses CCR5 as a coreceptor for entry into the cell. Virus may use CCR5 (R5-tropic) or CXCR4 (X4-tropic) of both (dual/mixed-tropic) coreceptors. The tropism test must be performed before therapy with maraviroc is initiated, as the drug is not effective against virus that is X4-tropic or dual/mixed tropic. Two tropism assays are commercially available: Trofile (Monogram Biosciences) and Sensitrop II HIV Co-Receptor Tropism Assay (Pathway Diagnostics, Malibu, Calif). The Trofile test is a cell-based assay in which pseudoviruses are constructed using the env gene sequence amplified from the patient’s plasma. This pseudovirus is then used to infect cells that express CXCR4 or CCR5. The tropism is determined according to which cell populations the pseudovirus infects.160 The Sensitrop assay uses a heteroduplex tracking assay (HTA) along with sequencing to identify minor viral populations that may be CXCR-4 tropic. HTA is performed using labeled probes with coreceptor-specific sequence combinations that are annealed to denatured PCR products derived from the total viral population and separated by gel electrophoresis. Mismatches with the probe result in altered migration of the heteroduplex and reveal distinct subpopulations of coreceptor-specific viral genomes. HSV causes both encephalitis and meningitis. In adults, HSV encephalitis is usually attributable to infection with HSV type 1, while HSV meningitis is most commonly caused by HSV type 2. HSV encephalitis is a severe infection with high morbidity and mortality; treatment with acyclovir reduces mortality from approximately 70% in untreated infection to 19 to 28%. Neurologic impairment is common (about 50%) in those who survive.161,162 HSV encephalitis may reflect primary infection or reactivation of latent infection. HSV meningitis is usually a self-limited disease that resolves over the course of several days without therapy. In some patients the disease may recur as a lymphocytic meningitis over a period of years.150,161 Neonatal HSV infection occurs in 1 : 3500 to 1 : 5000 deliveries in the United States.161 It is most commonly acquired by intrapartum contact with infected maternal genital secretions and is usually due to HSV type 2. In the newborn, three general presentations of the disease are known: (1) skin, eye, and mouth disease, which accounts for approximately 45% of infections; (2) encephalitis, which accounts for 35%; and (3) disseminated disease, 20%. Because disseminated disease is often associated with neurologic disease, CNS disease occurs in about 50% of newborns with neonatal HSV infection. HSV encephalitis cannot be distinguished clinically from encephalitis caused by other viruses such as West Nile Virus, St. Louis encephalitis virus, and Eastern equine encephalitis virus. Historically, the gold standard for the diagnosis of HSV encephalitis required brain biopsy with identification of HSV by cell culture or immunohistochemical staining. This approach provided high sensitivity (99%) and specificity (100%), but it required an invasive procedure, and several days elapsed before results were available. Cell culture of CSF has a sensitivity of less than 10% for the diagnosis of HSV encephalitis in adults. Tests that measure HSV antigen or antibody in CSF have diagnostic sensitivities of 75 to 85%, and diagnostic specificities of 60 to 90%.161 Because of the limitations of conventional methods, there was interest in assessing the clinical utility of PCR for the detection of HSV DNA from CSF of patients with encephalitis. The two largest studies compared HSV PCR on CSF specimens versus brain biopsy4,84 in patients with suspected HSV encephalitis. The sensitivity and specificity of PCR were greater than 95%, and the sensitivity of HSV PCR did not decrease significantly until 5 to 7 days after the start of therapy. PCR is positive early in the course of illness, usually within the first 24 hours of symptoms, and in some individuals, HSV DNA can persist in the CSF for weeks after therapy is initiated.4,84,163 The clinical utility of HSV PCR has also been established for the diagnosis of neonatal HSV infection. In one study,79 HSV DNA was detected in the CSF of 76% (26 of 34) of infants with CNS disease; 94% (13 of 14) of those with disseminated infection; and 24% (7 of 29) of infants with skin, eye, or mouth disease. The persistence of HSV DNA in the CSF of newborns for longer than 1 week after therapy is initiated is associated with a poor outcome.90 Based on these findings, detection of HSV DNA in CSF by PCR has become the standard of care for the diagnosis of HSV encephalitis and neonatal HSV infection. In newborns with disseminated disease, HSV DNA may be detected in serum or plasma specimens, and it can be a useful diagnostic tool in newborns if it is not possible to do a lumbar puncture. Although the sensitivity of HSV PCR is high, it is not 100%, so a negative PCR test may not rule out neurologic disease due to HSV, particularly if the pretest probability is high. In this situation, it is important to consider repeat testing. As with HSV encephalitis, HSV meningitis cannot be distinguished clinically from other viral meningitides, although recurrence of viral meningitis is a strong clue that HSV may be the etiologic agent. Unlike HSV encephalitis, HSV meningitis has not been the subject of large studies evaluating the clinical utility of PCR for diagnosis. Nonetheless, because the sensitivity of cell culture of CSF specimens is only 50%, HSV PCR of CSF is commonly used in the evaluation of meningitis and has been described as accurate in anecdotal reports.135,150 HSV PCR assays need low detection limits (several hundred copies/mL of specimen) to be useful in evaluating neurologic disease. This is particularly true for the diagnosis of meningitis, where CSF concentrations of DNA tend to be lower than those seen with encephalitis. HSV neurologic disease rarely occurs in individuals without an increased CSF white blood cell count or protein concentration.140 Caution should be exercised in applying this generalization to immunocompromised individuals, as they may not mount a typical inflammatory response to HSV infection. Although HSV PCR of CSF specimens is clearly the gold standard for the diagnosis of neurologic disease, results should be interpreted with caution because neither sensitivity nor specificity is 100%. Test results should always be interpreted within the context of the clinical presentation of the patient. If results do not correlate with the clinical impression, repeat testing should be performed.
Molecular Methods in Diagnosis and Monitoring of Infectious Diseases
Chlamydia Trachomatis and Neisseria Gonorrhoeae
Nucleic Acid Testing for CT and GC
False-Positive Test Results
False-Negative Test Results
Liquid Cytology Specimens
Recommendations on Laboratory Testing for CT and GC
Human Papillomavirus
Nucleic Acid Testing for HPV
Human Immunodeficiency Virus Type 1
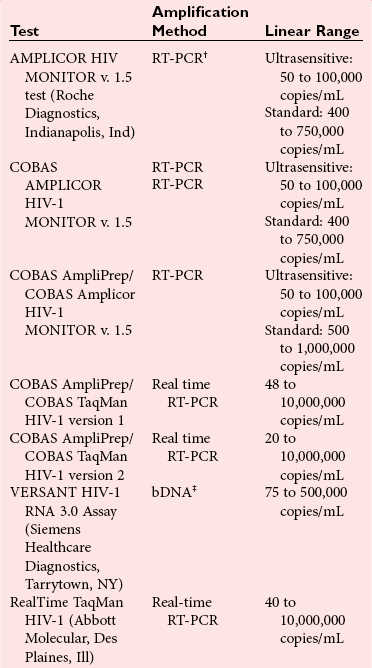
HIV-1 Viral Load Testing
Available HIV-1 Viral Load Assays
Qualitative and Proviral HIV-1 RNA Testing
HIV-1 Resistance Testing
HIV-1 Tropism Testing
Herpes Simplex Virus
Nucleic Acid Testing for HSV
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Basicmedical Key
Fastest Basicmedical Insight Engine
