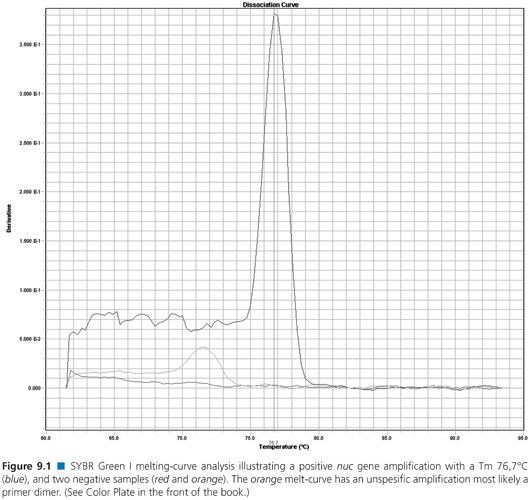
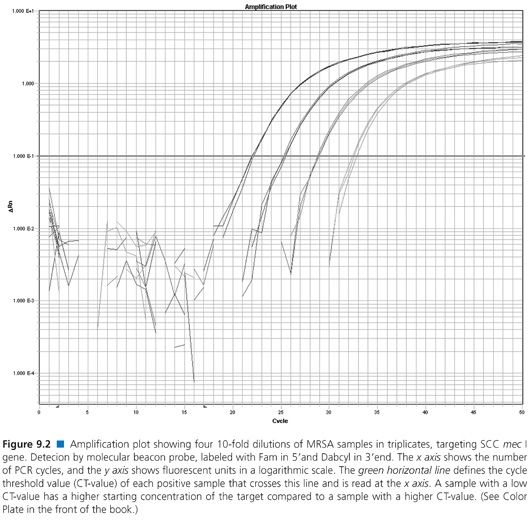
Table 9.1 descriptions include commonly used NAATs such as traditional polymerase chain reaction (PCR), multiplex PCR, real-time PCR, nucleic acid sequence-based amplification (NASBA), loop-mediated isothermal amplification (LAMP), and helicase-dependent amplification (HDA). Several detection systems used by real-time PCR are not mentioned, that is, Scorpions primers (Biosearch Technologies, Petaluma, CA), MGB Eclipse probes (EliTechGroup, Paris, France and Life Technologies, Grand Island, NY), Light-up probes (LightUp Technologies AB, Huddinge, Sweden), HyBeacon probes (Hain Lifescience GmbH, Nehren, Germany), Yin-yang probes (QuanDx, Menlo Park, CA), or Amplifluor (EMD Millipore, Billerica, MA). Examples of a real-time PCR melt curve and an amplification plot from real-time PCR probe detection are displayed in Figures 9.1 and 9.2, respectively.
Microarrays
The traditional Southern or Northern blot hybridization principles were used to develop microarrays for fast parallel, high-throughput detection and quantitation of nucleic acids (23). In nucleic acid arrays, hundreds or thousands of sequence-specific probes such as oligonucleotides or PCR products are deposited in fixed regions on a solid support or chip. Labelled targets applied to the solid support then hybridize to sequence-specific regions. The purposes for application of microarrays include large-scale genotyping (including single nucleotide polymorphism [SNP] analyses), gene expression profiling, and comparative genomic hybridizations (including copy number variation analysis). For a thorough introduction to microarray technology, including its applications, fabrication, target preparation, hybridization, detection, and data analysis, see Dufva (24). Both in-house and commercial microbial diagnostic microarrays have been developed to detect microbes as well as genes involved in antimicrobial resistance and virulence (25). The easy-to-use commercial microarray technology multiplexing power is attractive for antimicrobial resistance screening in central high-throughput facilities or hospital labs. A limitation of microarrays is that the signal does not provide any other information than the presence of the target. The size or complete sequence of the captured target molecule is not revealed.
The Application of Matrix-Assisted Laser Desorption-Ionization Time-of-Flight Mass Spectrometry Opens New Perspectives in the Detection of Antimicrobial Resistance
Bacterial identification based on mass spectral fingerprints obtained by MALDI-TOF MS is now routinely used in many clinical laboratories. The use of this technology in identification of antimicrobial resistance is rapidly increasing. For a detailed presentation of the use of MALDI-TOF MS in detection of antimicrobial resistance mechanisms, see Hrabak et al. (26). Briefly, antimicrobial resistance can either be revealed by changes in whole cell protein mass spectra when comparing susceptible and resistant isolates; by changes in mass spectra representing antimicrobials and their enzymatic degradation products or wild type and modified proteins/RNA; or by peptide, RNA, and DNA mini-sequencing (26–29).
The potential use of MALDI-TOF MS in detection of vancomycin-resistant enterococci (VRE) (30) and methicillin-resistant Staphylococcus aureus (MRSA) have been described but needs further refinements and validation (31). MALDI-TOF assays have also been developed to identify the enzymatic degradation of β-lactams by β-lactamases (32,33) or to reveal methylation of 23S rRNA by the cfr (chloramphenicol–florfenicol resistance) gene (34). Furthermore, MALDI-TOF MS–based mini-sequencing can reveal nucleotide polymorphisms responsible for antimicrobial resistance, that is, TEM-type extended-spectrum β-lactamase polymorphisms (35) and drug-resistant M. tuberculosis (36). Proteomic analyses using MALDI-TOF MS to identify proteins associated with resistance by peptide mass fingerprinting following sodium dodecyl sulfate polyacrylamide gel electrophoresis or 2D electrophoresis has been used to identify outer membrane or periplasmic proteins with different expression levels in resistant compared to susceptible isolates (37–41). Application of MALDI-TOF MS in proteomic analyses can complement molecular genetic techniques in reference or research laboratories. However, some of their applications will be challenged by the use of WGS in the near future (26).
Nucleotide Sequencing Techniques and Their Application in Detection of Antimicrobial Resistance
Traditional Sanger dideoxy terminator sequencing, the gold standard for 30 years, has mainly been used to identify SNPs resulting in antimicrobial resistance rather than determining the presence of acquired resistance genes. Once the acquired genetic trait has been explored, the presence or absence of this is sufficient to reveal resistance mechanisms. The expanding need for whole genome sequence information has fuelled a technical revolution. Today’s sequencers provided large volumes of genetic information in high resolution at dramatically decreased cost. Recent reviews have in detail described available first- to fourth-generation sequencing methods and their relevant characteristics and applications (42,43).
The turnaround time, cost, and user-friendliness of WGS have improved tremendously. Thus, whole bacterial genome sequencing is predicted to be a future routine method for species identification, the examination of antimicrobial resistance and virulence determinants as well as monitoring the evolution and clonal spread of bacterial pathogens (18).
Rapid WGS has already been used to investigate outbreaks of MRSA (44) and carbapenem-resistant Klebsiella pneumoniae (45) and to unravel the molecular mechanisms in development of antimicrobial resistance during antimicrobial treatment of infections with multidrug-resistant Acinetobacter baumannii (46) and K. pneumoniae (45). Zankari et al. (47) have developed a continuously updated Web-based program (ResFinder at http://www.genomicepidemiology.org) presenting an easy way to identify acquired antimicrobial resistance genes in whole genome data. However, it is important when using this database to adjust the software ID threshold to less than 100% to find resistance genes that have mutations not recorded in the database. Furthermore, the results are categorized into different antimicrobial groups, but it is still important to understand the resulting genetic information in the context of the antimicrobial susceptibility profile.
The use of sequencing in clinical microbiology also call for a quality standard for sequence-based assays defining DNA sequence quality as well as quality of reference sequences from external databases. Extended comments on automated sequence quality assessments and the quality control of currently available databases have been pertinently outlined by Underwood and Green (48).
MOLECULAR METHODS: QUALITY ASSURANCE
It is critical that the molecular assays are sufficiently validated and quality assured for diagnostic purposes. This is not obvious even for methods published in peer review journals (authors own experiences). The validation of an assay has some key parameters to test such as sensitivity, specificity, accuracy, precision, limit of detection, detection range, repeatability and reproducibility, and external quality assessment (EQA)/proficiency testing, for example, Quality Control for Molecular Diagnostics (http://www.qcmd.org) (49,50).
Nucleic Acid Extraction
High-quality RNA/DNA provided by nucleic acid extraction is a prerequisite for the performance of NAAT. Both in-house and commercial as well as manual, semi-automated, and fully automated extraction systems are used for nucleic acid extraction. Silica solid-phase extraction by centrifugation or vacuum, coated and uncoated magnetic beads extraction, and magnetic silica beads extraction are commonly used in automated systems. The four main steps in nucleic acid extraction are cell disruption, lysis, wash and removal of cell debris and inhibitor buffer, and, finally, recovery of pure nucleic acid without inhibitors.
Inhibitors and enhancers can influence the sensitivity of a NAAT-based method at all stages. Inhibitors can originate from the clinical sample or extraction buffers, typically the lysis buffer or ethanol. The inhibitors can be the result of suboptimal extraction protocols not suitable for the test material or difficult-to-treat clinical samples such as sputum or whole blood.
The credibility of the sample result of a NAAT depends on the validation process of the assay and on types and number of controls that runs every time the assay is performed. The controls are meant to assure you that the process has been optimally performed. During nucleic acid extraction, positive and negative process controls (PPC and NPC) are needed. The PPC is a known positive reference strain that is diluted to 1 log above the limit of detection. Transport medium or ddH2O is typically used as NPC. The purpose of the PPC is to confirm that the extraction process is performed optimally. If the PPC is negative after the NAAT, you need to rerun all samples from extraction. The purpose of the NPC is to check for cross-contamination during extraction.
An internal control (IC), also called internal amplification control (IAC) or internal positive control (IPC), is an inhibition control. The IC can be an oligo, plasmid or purified PCR product that will be amplified during the NAAT. Inhibition control is preferably introduced before the extraction step, for example, spiked in the magnetic beads solution, but can also be introduced from the amplification step, for example, spiked in the NAAT master mix. All negative samples should detect the IC; otherwise, the sample result should be discharged. The sample or nucleic acid elute should then be diluted 1:1 and rerun. When performing NAAT, it is recommended to include a positive and a negative amplification control.
METHICILLIN-RESISTANT STAPHYLOCOCCUS AUREUS—A TARGET FOR MOLECULAR DETECTION WITH INCREASING COMPLEXITY
Characteristics of Methicillin-Resistant Staphylococcus aureus
S. aureus is a major human pathogen within health care institutions and in the community. Thus, the overall resistance profile in clinical strains of S. aureus has an important impact on guidelines for empirical treatment of bacterial infections in general and bloodstream infection in particular. Wild-type S. aureus is naturally susceptible to β-lactam antimicrobials with the exception of monobactams. The high prevalence of penicillinase production in S. aureus has rendered the penicillinase-stable penicillins (oxacillin, cloxacillin, nafcillin, methicillin) the most active β-lactams.
Resistance toward penicillinase-stable penicillins in S. aureus is named methicillin resistance although this antimicrobial is not in clinical use. Methicillin resistance in S. aureus (MRSA) is caused by the acquisition of a mecA gene encoding a β-lactam low-affinity penicillin-binding protein (PBP), termed PBP2a or PBP2’. PBP2a substitute the essential functions of the high-affinity PBPs in the presence of β-lactam antimicrobials, rendering the bacteria resistant to this important class of antimicrobials (51). The regulation of mecA expression is complex. Expression of mecA in a minor fraction of the bacterial population (heterogeneous expression) challenge phenotypic detection and requires molecular confirmation (52).
The new cephalosporins (ceftobiprole and ceftaroline) have a strong affinity for PBP2a and are active against MRSA (53). However, penicillinase-stable penicillins remain the reference β-lactams in the treatment of S. aureus infections in the absence of methicillin resistance. Infections with MRSA are associated with higher morbidity and mortality and increased hospital costs (54). Thus, rapid and accurate detection of MRSA from pure culture or directly in clinical samples are important for tailoring individual antimicrobial treatment as well as the implementation of appropriate infection control measurements to prevent the spread in health care institutions. Molecular techniques are used to confirm phenotypically suspected MRSA and to reduce the detection time in clinical samples.
Molecular Methods for Detection of Methicillin-Resistant Staphylococcus aureus
The development of molecular methods for detection of MRSA has been based on our current understanding of the genetic support of the mecA gene and its wide distribution in S. aureus and several coagulase-negative staphylococci (CoNS). The conserved 2.1 kb mecA gene is carried by a mobile genetic element in staphylococci, designated the staphylococcal cassette chromosome mec (SCCmec) (55). The SSCmec varies in size and content. A number of SCCmec types have been characterized (http://www.sccmec.org/Pages/SCC_HomeEN.html) and more diversity is to be expected. SSCmec encode recombinases (cassette chromosome recombinases [ccr]) that mediate excision and site-specific integration at the chromosomal attB site near the 3′ end of orfX.
An array of different molecular strategies has been developed for the detection of MRSA, and selected publications are presented in Table 9.2. Some methods have targeted mecA itself and linked S. aureus–specific chromosomal genes (nuc, femA) in multiplex formats to differentiate between MRSA and other methicillin-resistant staphylococci (56–58). New insights into the detailed organization of SCCmec provided new concepts for MRSA-specific PCRs bridging the SSCmec-chromosomal orfX junction site (59). Developments of these strategies into real-time PCR assays have significantly improved their application in clinical microbiology. These methods are, together with the immunologic detection of PBP2’, used as standard tests for MRSA detection and confirmation.
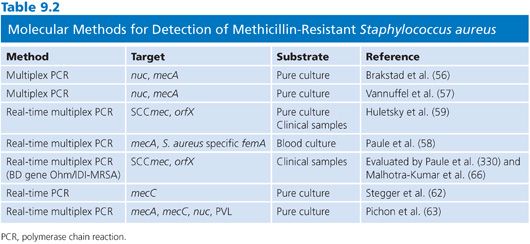
Challenges and Possibilities for Molecular Diagnostics of Methicillin-Resistant Staphylococcus aureus
The recent detection of a novel mecA homologue in human and bovine S. aureus populations has challenged our current molecular methods for detection of MRSA (60). The new mecA homologue is now classified as mecC in line with phylogenetic principles. mecC shows only 70% nucleotide sequence homology to mecA. Consequently, mecC and its corresponding PBP is not detected by conventional mecA-based PCRs or commercial immunologic PBP2’ assays, respectively (61). Recent progress in molecular detection of MRSA has taken our extended knowledge on mec-alleles into consideration, both in the design of new PCR formats (62,63) and in a rapid microarray-based detection (64).
The increasing repertoire of clinically relevant mec-homologues will also challenge the prospects for a rapid proteomic-based approach in the detection of MRSA. Both MALDI-TOF MS methods as well as surface-enhanced laser desorption-ionization time-of-flight (SELDI-TOF) MS has been described for the discrimination between methicillin-susceptible S. aureus (MSSA) and MRSA (31,65). A recent review on the application of MALDI-TOF MS in the detection of antimicrobial resistance concluded that the previously mentioned test needed further validation, although reproducible MRSA-specific profiles were obtained (26).
In conclusion, genetic detection of MRSA from pure cultures and complex clinical samples can be approached by a diversity of amplification-based techniques including in-house and commercial methods (66). Proteomic-based assays, except immunologic PBP2’-detection, need further refinements to be used in routine diagnostics. Methods based on the detection of SCCmec integration sites should take into account the increasing modular and sequence diversity in SCCmec types that may provide both false-positive and false-negative results. Moreover, antimicrobial selection will continue to facilitate transfer and stabilization of new mec homologues from commensals to S. aureus and challenge our molecular diagnostic repertoire targeting only known mec types (61).
DETECTION OF ACQUIRED GLYCOPEPTIDE RESISTANCE GENES
VRE and vancomycin-resistant Enterococcus faecium (VREfm) in particular are common nosocomial pathogens worldwide (67). The rates of vancomycin resistance monitored in medical centers in Europe, North and Latin America, and the Asia-Pacific region in 2004 to 2006 were less than 5% for Enterococcus faecalis and varied from 12% in Europe to 66% in North America for E. faecium (68). The implementation of measures to control health care–associated VREfm are by many considered unmanageable with those resistance rates. However, the burden of VRE infections should not be belittled (70). VRE bloodstream infections are associated with increased mortality. Increased density of van-determinants and associated resistances will raise the risk of transfer to other gram-positive pathogens.
The van Alphabet
The vancomycin resistance cluster (van) alphabet in enterococci currently consist of nine gene clusters, namely the acquired vanA, vanB, vanD, vanE, vanG, vanL (69), vanM (71), and vanN (72), and the intrinsic vanC genotype in Enterococcus gallinarum and Enterococcus casseliflavus. Resistance is due to synthesis of altered peptidoglycan precursors with peptide side chains that terminate in D-lactate (vanA, vanB, vanD and vanM) or D-serine (vanC, vanE, vanG, vanL, and vanN) for which vancomycin has lower affinity than the normal D-alanine side chain terminus (73–76). Their characteristics and species distribution are summarized in Table 9.3. Both vanA, vanB, vanG, vanM, and vanN have been shown to be transferable between enterococci as part of large conjugative chromosomal elements or plasmids (69,71,72). However, the vast majority of VRE is associated with vanA and vanB determinants hosted by E. faecium or E. faecalis. Importantly, several independent cases of vanA transfer to S. aureus and MRSA have been reported (77). S. aureus may coexist with VRE in the gastrointestinal tract or superficial wounds, providing a likely reservoir for development of vancomycin-resistant S. aureus (VRSA) (78).
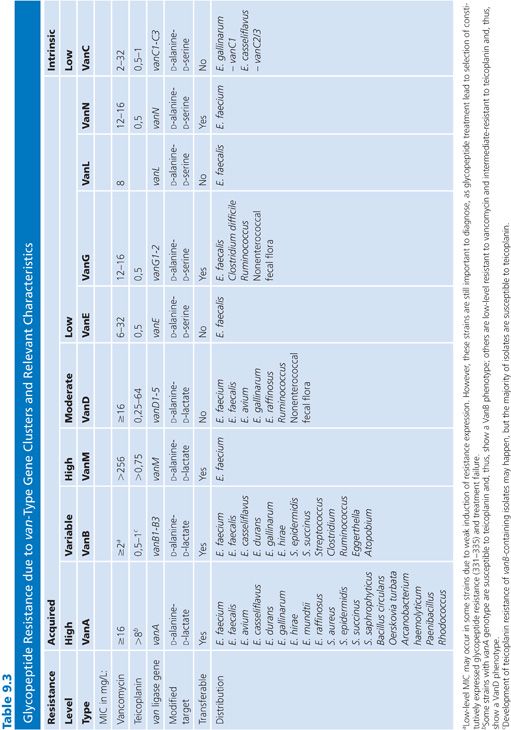
The vanA genotype is currently the most prevalent VRE genotype worldwide, but rates of infections with vanB-type VRE (mainly VREfm) infections are increasing in several European countries and are predominant in Australia (79–85). In contrast to vanA, the vanB ligase gene has been divided into three subtypes, vanB1–3, based on phylogenetic diversity (86–88). The most prevalent vanB2 subtype (89–100) has been identified in adapted bacterial genera of the normal intestinal flora such as Atopobium, Clostridium, Ruminococcus, Eggerthella, and Streptococcus (101–104). The vanB2 subtype has also been observed in two clinical samples of Atopobium and Clostridium (104). Interestingly, the vanB2 subtype has a high prevalence in community and hospital human fecal specimens in the absence of cultivable VRE (105–107). The high rates of nonenterococcal vanB in fecal samples result in a low predictive positive value for VRE using PCR detection of vanB directly from fecal samples (107–111), whereas vanA detection is more specific (111–113).
Possible Methods for Molecular Detection of van Genes
MALDI-TOF MS technology has been used to identify and investigate the epidemiology of an outbreak with vanB-positive VREfm (30), and an array of Raman-enhancing nanoparticles coated with vancomycin has been employed to capture VRE from human blood (114). Thus, the future holds promise for use of novel techniques to detect VRE. However, today, most described techniques for molecular detection of glycopeptide resistance involves amplification of the van genes.
A number of amplification techniques to detect the glycopeptide resistance genes have been developed. Methods that detect at least the two most common van genotypes (vanA and vanB) and evaluated on well-characterized strain collections or clinical specimens in comparison with in vitro susceptibility tests are listed in Table 9.4.
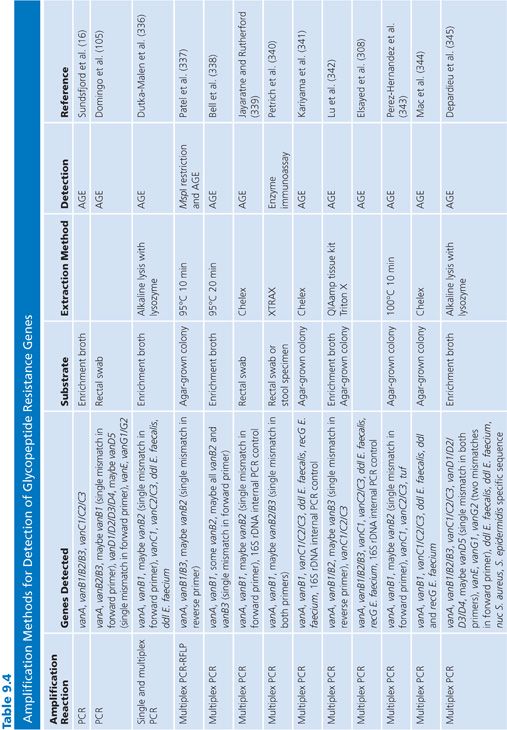
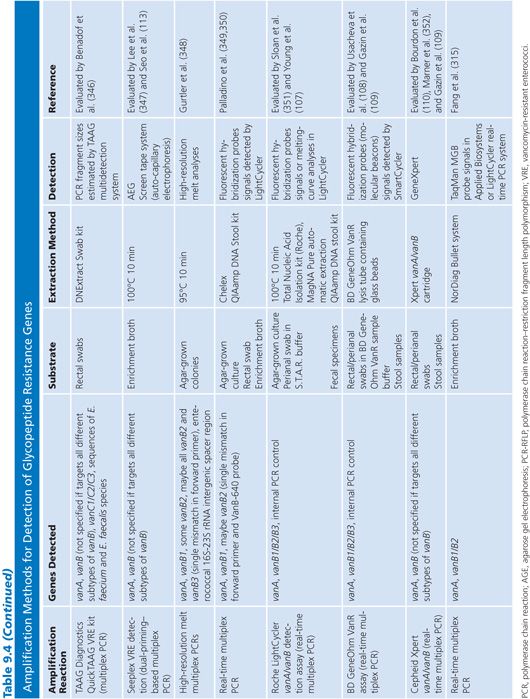
Amplification techniques are easy and useful tools for reference laboratories and routine labs that wish to confirm the phenotypic detection or investigate the molecular epidemiology of VRE. However, there are some aspects that are important to consider when applying these techniques. The vanB, vanD, and vanG determinants have subtypes. Thus, primers and probes described in the literature to hybridize to these van genotypes have been checked in silico against all subtypes to reveal ambiguities and notes of which subtypes they are likely to cover have been made in Table 9.4. Importantly, although the vanB2 subtype is the most widespread subtype of vanB, not all PCR-based methods detect this subtype.
Because the van genes can be found in other bacterial genera than enterococci, it is important to consider when performing screening on biologic samples that many molecular methods detect the van genes and not VRE per se. Methods that detect only the van genes are thus expected to have a higher rate of false-positive results. Some of the methods listed in Table 9.4 include species-specific (E. faecalis and E. faecium ddl, E. faecium recG) or genus-specific (tuf) genes for identification of enterococci or staphylococci (S. aureus nuc, Staphylococcus epidermidis species-specific gene) to overcome this problem.
An oligonucleotide DNA microarray with 105 probes that target 42 genes of the glycopeptide resistance gene clusters vanA, vanB, vanC, vanD, vanE, and vanG including the subtypes of vanB, vanD, and vanG as well as detection of ermB and the E. faecalis specific gene lsa correctly identified the different genotypes of reference and other VRE strains with various phenotypes (115). This microarray give additional and complementary information compared to phenotype alone and is suitable for reference laboratories that wish to reveal the genetic content of the van clusters explaining defective gene clusters and identifying additional resistance elements masked by a similar resistance phenotype. This array is not commercially available. Moreover, all presently known van clusters are not included in this array. Thus, WGS will be a better research approach to identify all present resistance elements contributing to a phenotype because this will also give the opportunity to explore the unknown.
RESISTANCE TO MACROLIDES, LINCOSAMIDES, AND STREPTOGRAMINS: RELEVANT RESISTANCE ELEMENTS, PHENOTYPIC EXPRESSION, AND THEIR DETECTION
Characteristics of Resistance to Macrolide, Lincosamide, and Streptogramin Antibiotics
A multiplicity of clinical relevant mechanisms of macrolide, lincosamide, and streptogramin (MLS antimicrobials) resistance is found (116–119). A recently updated database on MLS resistance genes is available at http://faculty.washington.edu/marilynr/. The nomenclature review of MLS resistance genes in 1999 defined that a new MLS gene must have a 79% or less amino acid identity with all previously characterized MLS determinants before receiving a unique name (116). The conservative criteria are controversial for some of the genes, as determinants with higher identity score may have different host range and genetic support (118,120). Herein, we focus on the most prevalent and clinical relevant determinants of resistance and their molecular detection. Phenotypic antimicrobial susceptibility testing distinguishes between the main groups of MLS resistance mechanism and guides antimicrobial therapy. Thus, the main purpose for genotypic identification is related to molecular epidemiology issues, understanding the dissemination and origins of MLS-related resistance determinants and their dynamics.
MLS antimicrobials have different chemical structures, but they share similar mechanisms of action. They are all inhibitors of bacterial translation. The structural analysis of bacterial ribosomal subunits in complex with antimicrobial inhibitors at an anatomic level has revealed their overlapping target sites (the peptidyl transfer center and the peptide exit tunnel) in the 50S subunit complex (121). Thus, they share many of the same resistance mechanisms (122).
The most commonly used macrolide antimicrobials include the natural product drug erythromycin and the semisynthetic derivatives clarithromycin and azithromycin. Macrolides are classified according to the number of atoms in the macrolactone ring: 14-membered (erythromycin and clarithromycin), 15-membered (azithromycin), and 16-membered (spiramycin). Ketolides (telithromycin) are semisynthetic derivatives of macrolides. Lincosamides (clindamycin and lincomycin) lacks the lactone ring. Streptogramin antimicrobials consist of a mixture of type A peptide–polyketide hybrids and type B cyclic depsipeptides that act synergistically by binding to the peptidyl transfer center and the peptide exit tunnel, respectively (5).
The spectrum of antibacterial activity of MLS antimicrobials is mainly restricted to gram-positive cocci (staphylococci and streptococci) and bacilli and gram-negative cocci. Gram-negative bacteria are, in general, inherently resistant to macrolides due to efflux mechanisms and drug inactivation. Exceptions include important human pathogens such as Bordetella pertussis, Campylobacter spp, Chlamydiales, Helicobacter pylori, and Legionella spp.
Mechanisms of Macrolide, Lincosamide, and Streptogramin Resistance and Their Genetic Determinants
There are three major mechanisms of MLS resistance: (a) target site modification by methylation or mutation that prevents the binding of the antimicrobial to its 50S ribosomal complex, (b) active efflux, and (c) enzyme-catalyzed drug inactivation. These mechanisms have been found in the bacteria producing these antimicrobials (5). The impact of the three mechanisms is unequal in terms of distribution and therapeutic implications in pathogenic bacteria. In large, modification of the ribosomal target complex confers broad-spectrum MLS resistance, whereas efflux and enzymatic inactivation affect only specific molecules. Resistance phenotypes affecting MLS antimicrobials and their genetic counterparts are presented in Table 9.5.
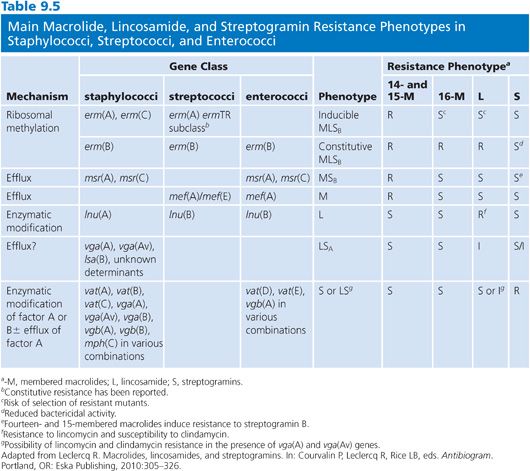
High-level resistance to macrolides is most often mediated by the enzyme-catalyzed methylation of the 23S rRNA by Erm (erythromycin ribosome methylase) methyltransferases encoded by erm genes that confer constitutive or inducible macrolide-lincosamide-streptogramin B (MLSB) resistance phenotypes (122). The number of described erm genes has already passed 40 (http://faculty.washington.edu/marilynr/). Four major erm classes are detected in pathogenic bacteria: erm(A), erm(B), erm(C), and erm(F) (118,122). Although erm(A) and erm(C) are typically detected in staphylococci, erm(B) class genes are mostly observed in streptococci and enterococci, whereas erm(F) are mostly described in Bacteroides and other anaerobes. In addition to the erm(B) genes, the ermTR genes (a subset of the erm[A] class) are widely distributed in β-hemolytic streptococci (118). The notion that each erm class may have a relatively specific distribution, but not strictly confined to a bacterial genus, reflects their association to mobile genetic elements and various mechanisms for horizontal gene transfer (118,123).
Low-level macrolide resistance is mainly associated with streptococci and linked to the production of an efflux pump (M phenotype) conferring resistance to erythromycin but not to clindamycin and/or streptogramins (122). A macrolide efflux system in streptococci was firmly established in 1996 (124). This system was phenotypically recognized and characterized to confer low-level resistance to 14- and 15-membered macrolides only. Several mef determinants have been described and the number varies dependent on criteria for definition of new elements (118,120).
A number of reports has shown marked differences between mef(A) and mef(E) (118). For instance, the different genetic elements carrying mef(A) or mef(E) and their contexts have been studied (125). Moreover, the two genes have disseminated markedly different in an ever growing number of species (120).
Molecular Detection Macrolide, Lincosamide, and Streptogramin Resistance Determinants
It is not much of a surprise to find that PCR is by far the most established method for the detection of MLS resistance genes and their genetic support. The high degree of similarity between major groups of resistance determinants does not allow a reliable discrimination to be made by using DNA hybridization experiments. Recommended primers for detection of MLS genes are available at http://faculty.washington.edu/marilynr/. Moreover, a diversity of different PCR primer combinations for amplification of the mef gene have been reported creating amplification products ranging from 202 to 1,759 bp (Table 3 in [120]). However, when using PCR to target resistance determinants with nucleotide diversity, one must take into account mismatches and in particular those located at the ultimate 3′-end of the primer sequence or multiple mismatches that may be present along the sequence of the primer(s). The use of such primers may result in an inefficient PCR or preferential amplification of mef gene subpopulations without the mismatch. A straightforward method for discrimination between mef(A) and mef(E) is based on the differential presence of restriction enzyme recognition sites in the two genes. Restriction enzyme digest resolved by agarose gel electrophoresis is sufficient to establish the difference (120).
The complexity of MLS-related resistance phenotypes are increasing. Several additional phenotypes affecting L or S antibiotics only (L phenotype) or in combination (LS phenotype) and their respective resistance determinants have been described lately (126–128). Importantly, lincosamide nucleotidyltransferases encoded by plasmid-mediated lnu genotypes and affecting the bactericidal activity of clindamycin have been described in staphylococci (117,122,127). Moreover, clinically relevant macrolide resistance in gram-negative bacilli has recently been described. Emerging plasmid-mediated mph(A) determinants encoding macrolide kinases was associated with azithromycin treatment failure in pediatric shigellosis (129).
In conclusion, the identification of MLS resistance mechanisms is important to guide the clinical use of MLS antimicrobials. The presence of various resistance determinants is highly dynamic and varies considerably between countries, species, and infections. Thus, it is important for empiric therapy guidelines that the phenotypic surveillance data on MLS susceptibility data are complemented with genotypic data showing the resistance determinants involved (122,130).
MOLECULAR DETECTION OF DRUG RESISTANCE IN MYCOBACTERIUM TUBERCULOSIS
The Problem
Drug-resistant M. tuberculosis (tuberculosis [TB]) represents a global public health threat (131). Diagnostic delays lead to increased mortality, selection for secondary resistance, and further transmission. Consequently, it is important to deliver reliable drug susceptibility test (DST) results in a clinically useful time frame.
Conventional phenotypic drug susceptibility testing of M. tuberculosis takes weeks to complete, although more rapid, commercialized, broth-based methods have been developed (132). Moreover, culture-based DSTs are labor-intensive and time-consuming and handling of TB cultures represents a potential biologic hazard. Thus, WHO has endorsed the development of molecular approaches to provide a rapid, relevant DST result to time-appropriate treatment.
Initial TB treatment is empirical, based on clinical suspicion and positive direct smears. Isoniazid (INH), rifampin (RMP), pyrazinamide (PZA), and ethambutol (EMB) are considered to be used as standard first-line treatment. Other antimicrobials including fluoroquinolones (FQs) are considered second-line drugs prescribed in case of resistance or intolerance. Multidrug-resistant TB (MDR-TB) is defined as resistant to at least the two main first-line anti-TB drugs, RMP and INH. In contrast, extensively drug-resistant TB (XDR-TB) is an MDR isolate that, in addition, express resistance to an FQ and at least one of the following second-line injectable drugs: amikacin (AMK), kanamycin (KAN), and capreomycin (CAP). The evolution of drug resistance in M. tuberculosis, including mechanisms and genetic regions involved in resistance formation has recently been reviewed (133).
Drug Resistance Mechanisms in Mycobacterium tuberculosis and Their Molecular Detection
M. tuberculosis is intrinsically resistant to a number of antimicrobials. This is partially due to a thick, lipid-rich cell wall that prevents antimicrobials for reaching their target as well as drug-inactivating enzymes such as β-lactamases (133). Thus, only a limited number of antimicrobials are effective treatment options. Acquired clinically relevant drug resistance in M. tuberculosis is not due to horizontal gene transfer. M. tuberculosis has not been shown to harbor plasmids. Further, the clonal population structure of TB indicates a limited role of horizontal gene transfer in the evolution of the species.
Hence, the genetic basis for acquired clinically relevant drug resistance in M. tuberculosis is chromosomal mutations that occur spontaneously during error-prone polymerization of DNA (134). Favorable mutations are selected for during antimicrobial selection and passed further through vertical transmission. The mutations are localized in genes for the antimicrobial target and affects drug–target affinity (RMP, FQs, aminoglycosides [AGs], and EMB). Mutations may also involve genes encoding drug-activation enzymes affecting the transformation of inactive antimicrobials to an active metabolite (INH, PZA, and para-amino salicylic acid [PAS]). A diverse array of commercial and in-house genotypic DSTs has been developed based on direct or indirect detection of resistance-conferring mutations of which some of them are selectively presented in Table 9.6 (135). Sequence- or hybridization-based detection of mutations in amplified products has become the standard techniques. Their success and limitations are partially based on the current knowledge of the factual mutations that account for phenotypic resistance (14).
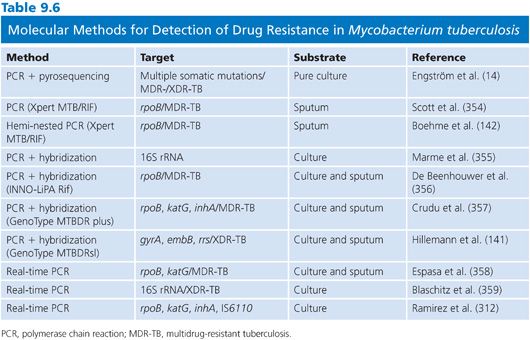
The mechanisms conferring resistance to the two main first-line drugs, RMP and INH, are the most relevant diagnostic targets in genotypic DSTs. RMP and/or INH resistance trigger therapeutic modifications, extended DST, and individual drug tailoring. Detection of MDR-TB requires the use of second-line drugs, and monoresistance leads to therapeutic adjustments in the consolidation phase to avoid unintended monotherapy and resistance development. RMP inhibits the early steps in transcription, binding the β-subunit encoded by rpoB (136). Phenotypic resistance is mostly confined to rpoB mutations in an 81-bp region named RMP resistance-determining region (RRDR). INH is a prodrug, activated by the catalase-peroxidase enzyme encoded by katG (137). Activated INH is believed to target a mycolic acid synthesis enzyme, enoyl-acyl carrier protein reductase InhA (138). Thus, phenotypic resistance to INH has been associated with mutations in katG affecting the activation of INH as well as inhA promoter mutations mediating InhA hyperexpression. Several in-house and commercial tests are available and proven effective in detection of both RMP and INH resistance including point-of-care diagnostics (139–141).
Although resistance mechanisms toward RMP, INH, and other anti-TB drugs are extensively investigated, they remain to be completely resolved. A recent comprehensive study of 290 clinical M. tuberculosis strains from four continents comparing phenotypic DST with pyrosequencing targeting mutations associated with resistance to RMP, INH, EMB, FQ, AG, and CAP showed a very high specificity in detection of MDR- and XDR-TB strains (14). However, the sensitivity of genotypic detection of phenotypic resistance to individual drugs varied considerably from 95% for RMP and 94% for INH to 61% for EMB, reflecting our incomplete understanding of clinically relevant resistance mechanisms.
In conclusion, specific and sensitive rapid commercial molecular tests for detection of both M. tuberculosis and RMP resistance directly from sputum samples have been validated and made available (142). The choice of molecular methods must take into consideration the local or national prevalence of M. tuberculosis and MDR-TB as well as available resources, technology, and competencies. Implementation of these technologies for routine point-of-care diagnosis of M. tuberculosis and MDR-TB at the primary care level is quite promising (143). Although feasible, high-burden countries are often associated with limitations in resources, personnel, and technology, restricting the use of these techniques to reference laboratories. Thus, to make a real impact in TB control, large scale implementation needs sustainable financial and operational support to meet the urgent need of cost-effective, simplistic methods for rapid, sensitive, and specific detection of M. tuberculosis and clinically relevant drug resistance outside reference centers. This is very important, as empiric treatment of TB without timely DST in regions with a burden of drug-resistant TB will continue to amplify drug-resistant TB at a high cost (144).
DETECTION AND IDENTIFICATION OF GENES CODING FOR EXTENDED-SPECTRUM β-LACTAMASES AND CARBAPENEMASES IN GRAM-NEGATIVE BACTERIA
The Growing Diversity of β-Lactamase
The first β-lactamase was identified as early as 1940 (145), before β-lactams were introduced into clinical use. Since then, the introduction of new β-lactam antibiotics in clinical practice and the corresponding emergence of new β-lactamases in human pathogens have evolved in parallel. Currently, the number of identified β-lactamases and variants has probably reached more than 1,000 (146) (http://www.lahey.org/Studies). In the last two decades, the dissemination and alarming prevalence of extended-spectrum β-lactamases (ESBLs) and carbapenemases have been the main concern. They are now threatening the use of β-lactams, which has been our largest and most valuable group of antimicrobials (147,148).
ESBLs are characterized by their hydrolytic activity against extended-spectrum cephalosporins (oxyimino-β-lactams) such as ceftazidime and cefotaxime, as well as being inhibited by the classical β-lactamase inhibitors (146). Carbapenems and cephamycins (e.g., cefoxitin) are generally not affected by ESBLs. The first ESBLs identified were variants of the TEM-1 and SHV-1 β-lactamases (149). The ESBL variants of TEM-1 and SHV-1 only differs from their progenitors by a few to a single amino acid change (149). The current number of TEM- and SHV-ESBL variants and the amino acid changes can be found on the β-lactamase Web site (http://www.lahey.org/Studies). In addition to TEM- and SHV-ESBL variants, the CTX-M ESBLs have emerged as the most prevalent ESBLs on a global scale (147,149–151). Currently, more than 130 CTX-M variants have been identified (http://www.lahey.org/Studies). Based on the amino acid sequence, the CTX-M ESBL can be grouped into five to seven subgroups/clusters: CTX-M-1/-3, CTX-M-2, CTX-M-8, CTX-M-9/-14, CTX-M-25, CTX-M-45, and CTX-M-64 (151–153). Other less prevalent ESBLs include GES, VEB, PER, BEL, BES, TLA, SFO, IBC, and OXA variants (149,154).
In contrast, carbapenemases are β-lactamases with the ability to hydrolyze carbapenems such as meropenem, imipenem, ertapenem, and doripenem (146). In addition, many of the carbapenemases have hydrolytic activity against all β-lactams. Consequently, the presence of one carbapenemase can give resistance to the whole β-lactam group of antimicrobials. Currently, the most significant problem is the global dissemination of mobile or acquired carbapenemases among various gram-negative bacteria (148,155).
Acquired carbapenemases have been identified in three Ambler classes of β-lactamases (A, B, and D). Acquired class A carbapenemases includes KPC, IMI, NMC-A, and SME where KPC is the most prevalent and 18 different variants (KPC-2 to KPC-19) have been identified so far (http://www.lahey.org/Studies) (148,155). The acquired class B carbapenemases includes the metallo-β-lactamases (MBLs) such as VIM, IMP, NDM, SPM, GIM, SIM, AIM, DIM, KHM, TMB, and SMB (148,155). The MBLs VIM, IMP, and NDM have shown a global dissemination and are the most prevalent MBLs (148,155). For these MBLs, several variants have also been identified to date with 41 VIM variants, 48 IMP variants, and 12 NDM variants identified to date (http://www.lahey.org/Studies). The acquired class D carbapenemases includes the OXA-carbapenemases, which can be grouped into two subgroups: (a) the OXA-carbapenemases, OXA-23–like, OXA-24/-40–like, OXA-58–like, and OXA-143, which are almost exclusively found among Acinetobacter spp and (b) the OXA-48–like carbapenemases found among Enterobacteriaceae (156,157). Although variable, all of these acquired carbapenemases have activity toward carbapenems. However, their activity toward other β-lactams varies. OXA-carbapenemases have limited or no activities toward extended-spectrum cephalosporins, and MBLs have no activities toward monobactams (aztreonam) (156,157).
Molecular Detection of β-Lactamases
Molecular methods for detection of ESBLs and carbapenemases are challenging for the clinical microbiology laboratory due to the large number of β-lactamase groups and diversity within groups. Further, the continuously changing epidemiologic landscape and emergence of new ESBLs or carbapenemases requires molecular methods to be adaptable to these changes. The presence of ESBLs and carbapenemases does not always result in clinical resistance according to the current breakpoints set by clinical breakpoints committees such as the European Committee for Antimicrobial Susceptibility Testing (EUCAST, http://www.eucast.org) or Clinical and Laboratory Standards Institute (CLSI, http://www.clsi.org). Further, it is advised that susceptibility results can be “reported as found” irrespective of the presence of an ESBL or carbapenemase. Consequently, molecular identification of ESBL or carbapenemase genes is not required for guidance of treatment. It should be noted that these guidelines are controversial and discussed (158).
Biochemical Methods for Detection of Extended-Spectrum β-Lactamases and Carbapenemases
The gold standard method for detection of β-lactamase activity is analysis of β-lactam hydrolysis by spectrophotometry. This method is mainly used in reference laboratories, as it requires specialized equipment and training in interpretation of the hydrolytic curves. Further, the method is relatively slow, as it requires overnight growth of liquid bacterial cultures and preparation of a protein extract (159,160). Recent evaluations of this method also indicate that detection of carbapenem hydrolysis by NDM and OXA-carbapenemases in A. baumannii is difficult (161).
Another, novel biochemical approach has been described for detection of ESBLs and carbapenemases where the hydrolysis of β-lactams is detected due to color changes of a pH indicator (phenol red) as the pH changes during hydrolysis (162–164). Inclusion of β-lactamase inhibitors to these assays also allows the discrimination between subclasses of carbapenemases (164). These novel assays can be implemented in clinical laboratories, as they are relatively easy to perform and bacterial colonies can be used, resulting in a total turnover time of less than 2 hours (162).
The availability of MALDI-TOF MS in microbiologic laboratories has led to the investigation into its possible application for detection of antimicrobial resistance (26). Several promising reports are now emerging where MALDI-TOF MS has been used for the identification of β-lactamase activity, particularly carbapenemase activity, by detecting degradation products of different β-lactams with good results (32,33,165,166). The use of MALDI-TOF MS for detection of carbapenemase activity has the potential to be implemented in a routine clinical microbiologic laboratory. However, as no specialized software or kits have been developed, expertise in manual interpretation of the spectra is required.
DNA-Based Molecular Methods for Detection of Extended-Spectrum β-Lactamases and Carbapenemases
Over the years, various molecular methods, including conventional PCRs, real-time PCRs, LAMP, and DNA microarrays, have been described for detection of ESBLs and carbapenemases. The majority of methods are developed by the scientific community and there are only a limited number of methods that are commercially available. The description of molecular assays in the following texts is an attempt to provide examples of methods that have been described and used and to provide a platform for selection of methods based on the possibilities in different laboratories.
An important aspect before selecting a molecular method for detection of ESBLs or carbapenemases is the purpose of the assay. The large number of genes/variants and the constant emergence of new ESBLs and carbapenemases make it difficult for clinical laboratories to be up-to-date. Specific assays are therefore limited to reference laboratories. In epidemiologic studies, it might be important to use assays that cover a broad range of genes while in outbreak settings, and for infection control purposes, more targeted specific molecular assays could be considered. From epidemiologic studies, it is known that the dissemination of ESBL and carbapenemase genes is often associated with specific bacterial clones such as Escherichia coli sequence type (ST) 131 and blaCTX-M, K. pneumoniae ST258 and blaKPC, and A. baumannii clones and blaOXA-carbapenemases (151,167,168). Molecular assays for rapid detection of some specific clones and associated ESBL or carbapenemase are developed, such as the detection of ST131 and blaCTX-M and ST258 and blaKPC (169,170).
Conventional and Real-Time Polymerase Chain Reactions for Detection of Extended-Spectrum β-Lactamases and Carbapenemases
Conventional PCR and real-time PCR are the most commonly used molecular methods for detection of ESBLs and carbapenemases. Numerous PCRs, including conventional single and multiplex PCRs as well as real-time PCRs, have been described (Tables 9.7 and 9.8). For the detection of TEM and SHV ESBLs and variants of GES with carbapenemase activity, the identification of mutations are required to distinguish from the progenitor variants that are not ESBLs or lack carbapenemase activity. In conventional PCRs, sequencing of the blaTEM or blaSHV PCR products and analysis for mutations in the sequence are required either through standard Sanger sequencing (171) or pyrosequencing (172). Real-time PCR methods for discrimination of SHV variants and non-ESBL variants have also been described using melting-curve analysis (173).
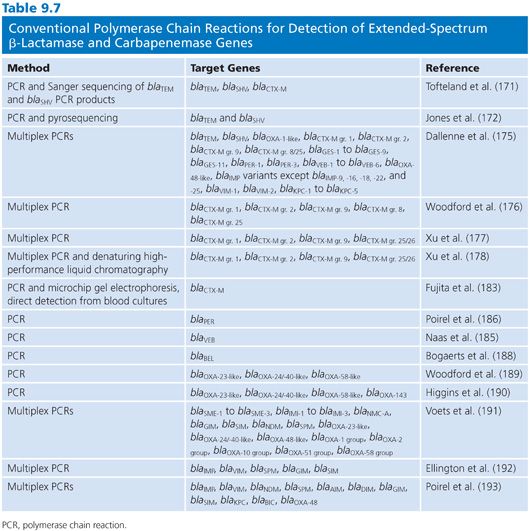
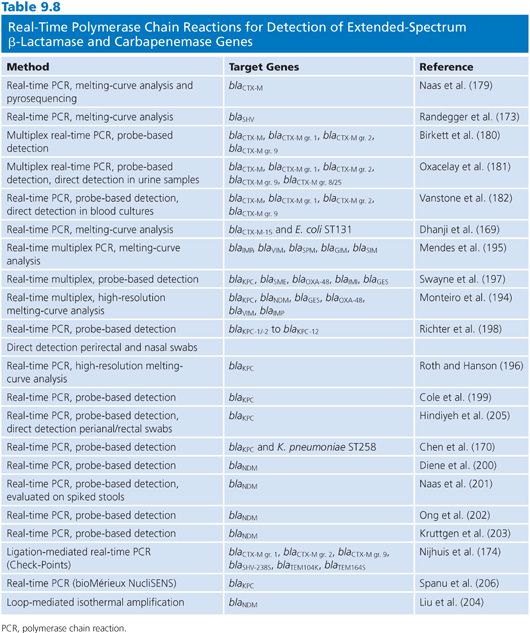
A commercially available ligation-mediated real-time PCR from Check-Points designed for the detection of specific common mutations of SHV and TEM ESBL variants as well as CTX-M have been evaluated (174). Although PCR detection of CTX-M do not require discrimination from a non-ESBL variant, it is often desirable to determine which subgroup the CTX-M variant belongs to. Multiple PCR assays including conventional PCRs (175–177), conventional PCR followed by denaturing high-performance liquid chromatography (178), real-time PCR followed by pyrosequencing (179), and real-time PCR using probe-based detection or melting-curve analysis (173,180–182) have been developed. Real-time PCR assays for direct detection of blaCTX-M in urine (181) and blood cultures (182,183) have also been reported. For the detection of the less prevalent ESBLs such as VEB (175,184,185), PER (175,184,186), GES (175,184,187), and BEL (188), conventional PCRs either as single or multiplex are mainly used for detection.
Various conventional PCRs and real-time PCRs have also been described for the detection of carbapenemase genes either as single or multiplex PCRs (Tables 9.7 and 9.8). There are also PCRs described which includes both detection of ESBLs and carbapenemases (175). Further, the developed PCRs vary with respect to the number of targets included. Conventional PCRs for detection of carbapenemases include a multiplex PCR for detection of the class D OXA-carbapenemases, blaOXA-23-like, blaOXA-24/-40-like, and blaOXA-58-like, mainly identified in Acinetobacter spp (189). This PCR has been updated with the addition of primers for blaOXA-143 (190). Further, different multiplex PCRs have been described for the detection of class A and class B carbapenemases (191–193).
With respect to real-time PCRs for carbapenemases, different detection methods such as melting-curve analysis (194–196) or fluorescent probes (170,197–203) have been developed (Table 9.8). For the detection of blaNDM, an assay using LAMP has been described with the potential of being a rapid test with low cost (204). Detection of blaNDM and blaKPC using real-time PCRs has been examined for their ability to detect these genes directly from clinical specimens such as perianal or rectal swabs (198,201,205). This can be particularly useful in outbreak settings or for screening of large populations. One commercially available real-time PCR assay from bioMérieux for the detection blaKPC has been evaluated by Spanu et al. (206).
Oligonucleotide Array–Based Technology for Detection of Extended-Spectrum β-Lactamases and Carbapenemases
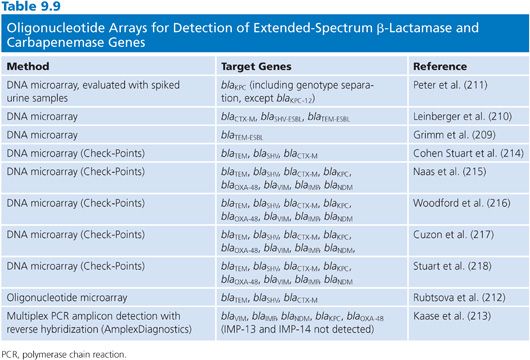
DNA microarray or oligo-based technology has the potential for detection of a large number of resistance genes and discrimination of variants within each class or family in a single test. In-house microarray assays have been developed for the detection of various resistance genes that also includes β-lactamases (207,208). However, the number of specific ESBL and carbapenemase genes in these arrays is limited. For the specific detection of ESBLs and carbapenemases, different approaches to this technology has been developed (Table 9.9). Grimm et al. (209) developed a DNA microarray for the detection of TEM ESBLs using Cy-labelled PCR products followed by hybridization on glass slides containing oligonucleotide probes. A further development of this assay has been done to include detection of SHV ESBL and CTX-M genes (210). A specific microarray for the direct detection and genotyping of blaKPC variants also using Cy labelling and glass slides has been reported (211). This assay has also been evaluated using spiked urine samples.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


