Resistance to β-lactam antibiotics can occur by one of three major mechanisms: inactivating enzymes (BLAs), low-affinity penicillin-binding proteins (PBPs), or decreased permeability. BLA is by far the most common mechanism associated with resistance to β-lactam antibiotics. The most common BLAs found among Bacteroides sp and Prevotella sp are cephalosporinases of the type 2e class. These BLAs are inhibited by sulbactam, clavulanic acid, or tazobactam, thus the increased potency of the β-lactam–BLA inhibitor combinations. Cefoxitin- and cefotaxime-inactivating enzymes and other BLAs have also been reported in many B. fragilis group species (60). The most potent BLAs are the zinc metalloenzymes encoded by either ccrA or cfiA genes of the B. fragilis group (61). These enzymes are responsible for the rare resistance to carbapenems, are active against all β-lactam antibiotics with known activity against anaerobes, and are not inactivated by current BLA inhibitors. Although resistance to carbapenems is currently quite rare in the United States, up to 3% of Bacteroides strains have been found to carry one of the genes expressed at a very low level. These strains can be induced to a higher level of resistance in the laboratory under selective pressure caused by a promoter (contained in an insertion sequence) that has inserted upstream of the ccrA or cfiA genes (29,62).
Production of BLAs by other anaerobic bacteria has been generally less well studied, but strains of Clostridium, Porphyromonas, and Fusobacterium organisms express resistance by one or more of these enzymes. Penicillin-resistant Fusobacterium and Clostridium organisms express penicillinases that are typically inhibited by clavulanic acid, although exceptions among some Clostridium sp have been reported (56,63).
Other mechanisms of resistance to β-lactam antibiotics are far less frequent in occurrence and less well studied. Decreased binding to PBP2 or PBP1 complex has been reported in rare clinical isolates in cefoxitin resistance among B. fragilis strains (64). Alterations in pore-forming proteins of gram-negative anaerobes are a third type of resistance, with the absence of one or more outer membrane proteins associated with high MICs to ampicillin/sulbactam in some strains of B. fragilis (65,66).
Metronidazole resistance occurs by the lack of reduction to its active form in anaerobic bacteria. Metronidazole-resistant B. fragilis group organisms, although rare, carry one of six known nim genes that appear to encode a nitroimidazole reductase that converts 4- or 5-nitroimidazole to 4- or 5-aminoimidazole, preventing the formation of the toxic drug form necessary for the agents’ activity (67,68). These genes have been identified on both the chromosome and on transferable plasmids. High-level expression of the nim genes requires an insertion sequence with a promoter, similar to that of carbapenem resistance (69). Differential gene expression affecting cell metabolism in Bacteroides has also been reported as an alternative mechanism for resistance (70). In contrast to Bacteroides, the mechanism of resistance to metronidazole for non-Bacteroides anaerobes is currently not known. Interestingly, metronidazole resistance in the microaerophilic organism Helicobacter pylori has been partially solved for some strains that contain mutations in the rdxA gene, an oxygen-insensitive nitroreductase that converts metronidazole to its active form in this organism (71). Other candidate genes for resistance include the flavin oxidoreductase (frxA), ferredoxin-like proteins (fdxA, fdxB), and pyruvate oxidoreductase (porA, porB) (72). Metronidazole also has activity against Mycobacterium tuberculosis, although apparently only in dormant cells, when reduction of the drug can occur. Resistance among actively growing Mycobacterium organisms is presumed secondary to a lack of sufficient reducing potential (73).
Fluoroquinolone resistance among Bacteroides sp has been attributed to either a mutation in the quinolone resistance–determining region of the gyrase A gene (gyrA) from single or multiple mutations, or an alteration in efflux of the antibiotic (74–77). High-level resistance may be secondary to both mechanisms in the same cell, although only a few strains have been tested to date. Both of these mechanisms appear to be responsible for the cross-class resistance to newer quinolones.
The lack of activity of aminoglycosides against anaerobes is related to the lack of uptake by the bacteria under anaerobic conditions and a failure to reach their ribosome targets (39). Tetracycline resistance is widespread, especially among B. fragilis group and Prevotella sp (15). Several genes encoding resistance have been identified among various anaerobes, which encode protective proteins, resulting in protection of the ribosomes. More importantly, however, is the association of tetracycline resistance and the inducible transfer of this resistance determinant upon exposure to low levels of the antibiotic. Chloramphenicol resistance is extremely rare but, when found, is associated with inactivation of the drug by nitroreduction or acetyltransferase (78).
METHODS FOR ANTIMICROBIAL SUSCEPTIBILITY TESTING OF ANAEROBIC BACTERIA
Several different methods, spanning five decades, have been utilized in antimicrobial susceptibility testing (AST) of anaerobic bacteria. During that time, more than 16 methods, 16 different media, and a host of other variables have been described to test susceptibility of anaerobes. NCCLS took the lead in developing a consensus for AST of anaerobes starting with the first approved standard in 1985, along with alternative methods published in a second document the same year. Subsequently, several revisions have been published that modified, added, or eliminated some methods (10). Following extensive multilaboratory collaborative studies sanctioned by the NCCLS in the late 1990s, a consensus was reached culminating in a single agar dilution standard and one broth microdilution method, both using the same medium (10,17,18). In addition to NCCLS methods, a very useful and highly correlated user-friendly method, Etest (AB Biodisk), has been FDA approved and available for several years (19,79). As a proprietary product, the Etest is not included in the CLSI documents. Of note, all methods can be performed in ambient air but require incubation in an anaerobic jar or glove box.
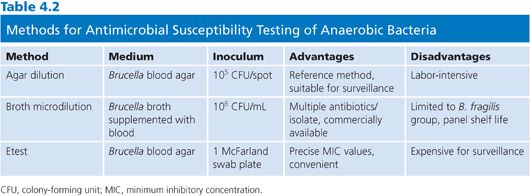
Choosing a method from among these three recognized and approved methods may depend on a number of factors (Table 4.2). The agar dilution standard has a very high degree of reproducibility but is fairly labor-intensive. A laboratory can test up to 30 isolates plus two controls, making it useful for batch testing. However, individual sets of dilution plates must be pored for each antibiotic, increasing material and labor costs. As the reference standard, agar dilution is most often used for evaluation of new antimicrobial agents. The broth microdilution method is more user-friendly than agar dilution and has the flexibility to test multiple antibiotics using the same microtiter plate, albeit only one isolate at a time. Based on available comparative studies of broth microdilution to the standard, results are considered equivalent when testing members of the B. fragilis group. However, results are not as comparable for non-Bacteroides anaerobes because of poor growth, and broth microdilution is not used for this group at this time. It should be noted that previously published studies testing non-Bacteroides anaerobes using a different broth (anaerobe MIC broth) were able to grow non-Bacteroides anaerobes with MIC results within twofold of those for agar dilution. Thus, broth microdilution testing for non-Bacteroides anaerobes can be considered but only if correlated with the current agar dilution standard. The third method, Etest, is relatively easy to perform and is well suited for testing individual isolates of any anaerobe. For surveillance testing, costs could be prohibitive.
CLINICAL AND LABORATORY STANDARDS INSTITUTE REFERENCE AGAR DILUTION METHOD
For the agar dilution reference standard, each test concentration of an antibiotic is mixed into molten agar and poured into separate Petri dishes to which an inoculum of an organism is applied, incubated anaerobically, and examined to determine the MIC for the antimicrobial agent tested. Methods described here are adapted from those of CLSI standards (10). For laboratories inexperienced with this method and anaerobe AST, a useful time table for setup and testing is provided as an appendix in the CLSI standards document.
Testing Medium
Brucella blood agar supplemented with 5 µg hemin, 1% vitamin K1, and 5% laked sheep blood is the recommended testing medium (10). Brucella agar blanks (17 mL) can be prepared in advance containing hemin and vitamin K1, and flash-autoclaved or microwaved and placed in an approximately 50°C water bath on the day of use (10). One milliliter of laked sheep blood is then added to each melted blank while still in the water bath. Two milliliters of each twofold diluted test antibiotic is then added to the agar, mixed by inverting the tubes, and poured into sterile Petri dishes (10). Following hardening of the plates, and drying briefly in an inverted position in a 37°C incubator, the plates are ready for use. Preferably, plates should be made on the day of testing but can be sealed in plastic bags and stored at 2°C to 8°C for periods of up to 72 hours if necessary (10). Exceptions to storage include plates containing clavulanic acid or imipenem/cilastatin, which must be made on the day of use.
Inoculum Preparation
The inoculum can be prepared by either a direct colony suspension or growth method. Direct colony suspension requires 24- to 48-hour growth on a Brucella blood agar plate (10). Several colonies are touched lightly with an inoculating needle or cotton swab and suspended in reduced Brucella broth to achieve a turbidity equivalent to a McFarland standard of 0.5 (10). The alternative growth method involves the inoculation of enriched thioglycollate medium (without indicator) with portions of five or more colonies from a Brucella blood agar plate, incubating for 6 to 24 hours at 37°C, and adjusting the turbidity to a McFarland standard of 0.5 by addition of reduced Brucella broth (10).
Inoculation and Incubation of Plates
Once the inoculum is prepared, it is most often applied using an inoculum-replicating apparatus, such as a Steers-Foltz replicator, to deliver 1 to 2 µL on the agar surface, corresponding to 1 × 105 colony-forming units (CFUs) per spot. Depending on the device, either 32 or 36 wells can be filled with different test organisms and controls using a Pasteur or other pipette (10). Application of inoculum to plates includes repeated stamping of plates, starting with lowest to highest dilution of each antibiotic set. One plate of supplemented Brucella blood agar without antibiotic should be stamped prior to and after each set of antibiotics for growth control. Contamination by aerobic bacteria during the inoculation procedure can be detected by inoculating a drug-free plate and incubated aerobically. Once plates are inoculated, they should sit until liquid is absorbed into the medium and then incubated in an anaerobic environment at 35°C to 37°C for 42 to 48 hours (10).
Interpretation of Results
End points are determined by reading each plate against a dark, nonreflecting background and comparing it with the control growth plate. Any growth on the aerobic control should eliminate further interpretation of that test organism. The end point for a given test organism is where a marked reduction occurs in the appearance of growth compared with control. A marked change includes a haze, multiple tiny colonies, or one to several normal-sized colonies. These descriptions have been problematic for those inexperienced with using this method. To that end, CLSI recommends the use of two figures containing 28 full-dilution color photographic examples of end point readings to illustrate the written descriptions (10).
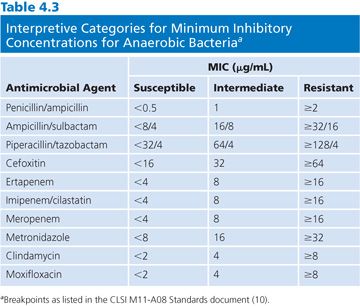
Interpretative categories approved by CLSI for MICs derived for anaerobic bacteria are shown in Table 4.3. This table includes agents that are the most frequently used in the clinical setting and were updated through 2012. Interpretative categories for any organism have been determined based on the population distribution of the bacteria, the pharmacokinetics, and pharmacodynamic properties of the antibiotic with verification of efficacy by clinical studies (see an in-depth description in Chapter 1). This works particularly well for single-organism infections. However, this is rarely the case for anaerobic bacteria, which are typically isolated from mixed infections. Many of the published anaerobic breakpoints were determined on the basis of animal models or the result of clinical trials involving patients with polymicrobial infections as well as pharmacokinetic data. Despite these potential limitations, the use of maximum dosages of antibiotics along with appropriate ancillary therapy (debridement or drainage) should be effective for organisms with susceptible breakpoints, although those with intermediate susceptibilities should be monitored closely (10).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


