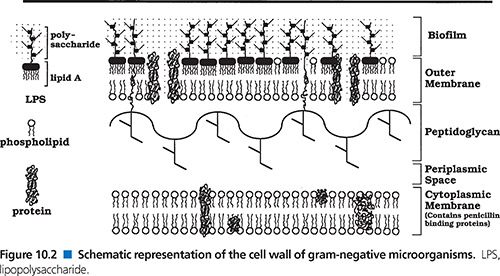
The cytoplasmic membrane in each group of bacteria is a semipermeable membrane that regulates molecular flow, in turn determining pH (143,148,149), osmotic pressures (150), and availability of essential substances. The peptidoglycan layer in each group is a continuous cross-linked mesh that forms a polyionic and amphoteric network (33,34,143). The peptidoglycan mesh is composed of linear glycan chains that are interlinked by short peptides (33,34,82,143,144,151). This shell surrounds the entire microorganism, is known as a sacculus, and is found exclusively in eubacteria. The peptidoglycan sacculus is not a rigid shell but instead is elastic and flexible. This relatively porous peptidoglycan sacculus (exclusion limits of 100,000 Da) serves as a mechanical “exoskeleton” that helps to maintain the microorganism’s shape, rigidity, and osmotic stability. The exoskeleton of gram-positive bacteria is thicker than that of gram-negatives, thus providing more rigidity. Although this polyanionic sacculus might appear to be an exclusion barrier, the exclusion limits of 100,000 Da make this meshwork very coarse and thus allow molecules of lesser size, such as antimicrobial agents, which have sizes of 300 to 700 Da, to readily diffuse through the layer. Finally, the peptidoglycan structure is involved in the cell division process (32,34,152).
The molecular structures of the cytoplasmic membranes of gram-positive and gram-negative bacteria are essentially the same, consisting of lipid bilayers containing phospholipids and membrane proteins (143–145). There are, however, important differences in the peptidoglycan wall and the biofilm for these two groups of bacteria. Gram-positive bacteria have a relatively simple but thick cell wall constructed of peptidoglycan and teichoic acids, which are long-chain polymers consisting of glycerol or ribitol residues with phosphodiester links and various substituents such as uronic acids (85,144,153). Teichoic acids are found as either cell-bound or free soluble acids. The cell wall of gram-positive bacteria contains two forms of the cell-bound teichoic acid. In one form, lipoteichoic acid, one end of the chain is anchored to phospholipids in the cytoplasmic membrane while the other end transverses the peptidoglycan layer in such a way that it protrudes at the cell surface (153). In the second, a cell wall teichoic acid, one end is attached to N-acetylmuramyl residues in the peptidoglycan layer and the free end protrudes at the cell surface. Finally, free, soluble teichoic acid is present in large amounts in the outer portion of the cell wall.
At the surface of the gram-positive cell wall, the protruding teichoic acids can be linked with one another via branching polysaccharides to form a matrix. The cross-linking of the biofilm matrix is accomplished using polysaccharides with repeating units of two or three sugars. The variety of possible hexose stereoisomers and of linkages, as well as the potential incorporation of unusual sugar residues, results in thousands of different trisaccharides in the matrix. Because the polysaccharide chains in the matrix are hydrophilic, water is absorbed into the matrix and transforms this outer layer into a gel. This matrix-supported gel (99% water) is known by a variety of names, including biofilm, glycocalyx, slime, alginate, and capsule (52,56).
One of the key components of the biofilm is phospholipids because of the covalent bonding they provide. In addition, this hydrated matrix depends on calcium and magnesium cations to maintain the negatively charged ends of the polysaccharides in close approximation. The availability of phospholipids and divalent cations in the medium greatly influences the final composition of the cell wall. This can be appreciated by considering the gram-positive cell wall. Under conditions of magnesium limitation, gram-positive bacteria increase the amount of teichoic acid produced while decreasing the amount of alanyl ester substitutions, which results in fewer polysaccharides present in the matrix and hence less biofilm. Phosphate limitation results in the replacement of teichoic acids by teichuronic acids, which have less covalent bonding. Starvation conditions, then, would be predicted to result in cell walls with minimal biofilm.
The cell walls of gram-negative bacteria differ from those of gram-positive bacteria in a number of ways (82,147,154). First, they have a relatively thin peptidoglycan layer, which provides less rigidity. Gram-negative cell walls also have an additional outer cell membrane (145–147) that serves as an effective permeability barrier (155–157). This outer cell membrane is composed of two layers. The inner layer consists largely of glycerophospholipid molecules with two covalently linked fatty acid chains, a molecular structure that is fairly common in membranes in general. The outer layer is somewhat unique among membranes and contains mainly two classes of proteins, lipoproteins and β-barrel proteins. Lipoproteins contain lipid molecules that are coupled with an amino-terminal cystein, whereas β-barrel proteins are β sheets wrapped into cylinders and referred to as outer membrane proteins (OMPs). The outer membrane also contains glycopeptides, principally lipopolysaccharides with six or seven covalently linked fatty acid chains. The fatty acids present in the lipopolysaccharides are saturated. This results in the interior of the lipid bilayer being less fluid, because there is no packing of carbons, as seen when the fatty acids are unsaturated (156). This serves to make this barrier more restrictive to hydrophilic agents.
Areas of adhesion between the outer cell membrane and the inner cytoplasmic membrane, called Bayer’s junctions, have been described by a number of investigators. These adhesions of the inner cytoplasmic membrane to the cell peptidoglycan/outer membrane are probable sites of synthesis of the outer membrane and biofilm as well as synthesis of other substrates to be pumped from the cell cytoplasm into the periplasmic space or from the cell cytoplasm directly into the biofilm.
The periplasmic space is an aqueous cellular compartment delineated by the outer cell membrane and the inner cytoplasmic membrane (158). The periplasmic space contains various proteins that are densely packed, making this aqueous milieu more viscous than the cytoplasm. These proteins include potentially harmful proteins such as RNAse or alkaline phosphatase, thus making the periplasmic space an evolutionary precursor of the lysosomes of eukaryotic cells (159).
Gram-negative bacteria, like gram-positive bacteria, are surrounded by a polyanionic polysaccharide matrix, which differs mainly by being anchored by the lipooligosaccharides rather than by lipoteichoic acids. In some instances, the attachment is to the peptidoglycan layer, whereas in others the lipid A is anchored to the inner cytoplasmic membrane. This lipopolysaccharide matrix of gram-negative bacteria is also thought to provide some selectivity/hindrance via negative ionic charges and/or steric hindrance (160).
Biofilms of both gram-positive and gram-negative bacteria are essentially anionic polymeric diffusion barriers and can be thought of as an ion-exchange resin of almost infinite surface area. In addition, the biofilm protects the microorganism from heavy metal toxicity, from most bacteriophages, from phagocytic white blood cells, from antibodies and/or complement, and from an inhospitable milieu such as osmotic, pH, or enzymatic dangers. The interface of the biofilm with the aqueous phase can be altered by methylation of fatty acids or by sulfonation of polysaccharides, which increases the barrier to water-soluble agents (161). Bacteria are able to excrete into their biofilm and the surrounding medium several different classes of molecules, including exopolysaccharides (the building blocks of biofilm), siderophores, protein enzymes, and toxins (146). Biofilms also appear to serve as a repository for defensive substances such as β-lactamase (162).
In human infections, microbial cells are most often found with biofilm (52,57,58,63), even though microscopic examination may not readily reveal this. In clinical microbiology laboratories, the optimal growth conditions sought by microbiologists are far from the near-starvation circumstances the bacteria encounter in their natural environments. This is particularly true for broth media (29). However, the growth of gram-positive and gram-negative bacteria on agar plates in the laboratory may reflect, in part, the presence or absence of biofilm. Smooth colonies have more biofilm than do rough colonies, while mucoid or slimy colonies have the most. Deep rough colonies, on the other hand, have the smallest amount of biofilm, if any at all. These deep rough mutants have most of the core lipid A eliminated. Such strains are more susceptible to lysozyme and more permeable to hydrophobic antibiotics. Finally, in clinical microbiology laboratories, biofilm may be recognized and described microscopically as capsule.
Consequences of Biofilm Disruption
The disruption of the bacterial cell wall often results in the death of the microorganism (114). This is well appreciated by clinicians. Less well appreciated is that the disruption of biofilm surrounding an individual microbial cell is not without consequences (163–165). These may be related to osmotic pressure and the shifting of cell peptidoglycan by that pressure. There is a higher hydrostatic pressure within the cytoplasmic space of a microbe than that which is exerted on the cell by the external milieu. The presence of the biofilm matrix seems to assist in keeping this internal pressure in check (172). When a portion of the gel is removed, however, the internal pressure shifts the cell wall/membrane so that it protrudes through this disrupted area, resulting in a fingerlike projection containing cytoplasmic contents (164,166–168). Extrusion of this portion of the cell wall/membrane through the hole in the biofilm is a result of the cell wall/membrane shifting to adjust to the focused pressure directed at the area of disrupted biofilm. This shift in turn activates autolytic enzymes to dissolve the peptidoglycan component of the cell wall as the wall shifts during replication (169). This causes dissolution of cell wall peptidoglycan in this area, which, in conjunction with the high, focused internal pressure, effectively severs this protruding bleb (170), leaving a transient hole. If enough holes are formed, leakage of cytoplasmic contents results in cellular death (171). The results of this process can be demonstrated by electron microscopy, which makes visible a range of ultrastructural changes, including narrow fingerlike projections, blebs, and extracellular cytoplasmic-filled vesicles. Disruption of the entire biofilm matrix, in contrast, tends to equalize the pressure over the entire cell wall/membrane. Consequently, when the autolytic enzymes dissolve the peptidoglycan of the entire cell, the result may be lysis of the entire cell or the creation of a spheroplast if the osmolarity of the external milieu is sufficiently high. When lysis is seen, it occurs rapidly, in contrast to the lysis seen after exposure to penicillin, where cells continue to grow for approximately half a generation before lysis (27,170). Finally, it appears that gram-negative bacteria are more susceptible to the effects of biofilm disrupters, perhaps because of their less rigid cell walls.
Disruption of biofilm can be accomplished by a number of physicochemical mechanisms (161). This disruption can be best appreciated by electron microscopy. The ultrastructure of normal cells of gram-negative or gram-positive bacteria has a slightly undulating smooth surface, which is transformed to a surface with blebs and tubular projections after displacement of Ca2+ and Mg2+ from the biofilm. Displacement of these cations from the biofilm by chelating agents such as ethylenediaminetetraacetic acid (EDTA) (155,170) or by polycationic agents (164,172) such as polymyxin B (166,167) and aminoglycosides (173,174) has been shown to be an effective way to disrupt biofilm, although this mechanism can be countered by the presence of excessive amounts of calcium and magnesium cations in the milieu (175–177). However, adding these cations after the damage has been done has no effect. The ultrastructural changes seen by electron microscopy are accompanied by a functional change, namely, increased permeability to hydrophobic agents such as antibiotics (146,163,178).
If the changes induced by the disruption of biofilm are not rapidly fatal and the cells are allowed to grow, the permeability barrier is repaired in about two-thirds of a generation (129). The addition of chloramphenicol or tetracycline or the omission of required amino acids does not affect the repair rate. A proton pump inhibitor such as 2,4-dinitrophenol, however, prevents repair. The activity of omeprazole and lansoprazole against H. pylori (179) may be related to their inhibitory effect on certain cellular membrane pumps. Omitting glucose also prevents biofilm repair. Interestingly, the addition of macrolides decreases the repair rate, possibly because of its inhibition of mRNA translation (180) as well as by the elaboration of exotoxins by means of codon–anticodon interactions that inhibit the translation of mRNA for inducible enzymes (130–138).
Effects of Antimicrobial Agents on the Production of Biofilm
Antimicrobial agents, not unexpectedly, can either increase or decrease the production of biofilm. This effect, in part, appears species-specific. For example, fluoroquinolones at concentrations of one-half the MIC of Staphylococcus epidermidis decrease the production of slime (i.e., biofilm) (181). On the other hand, exposure of Klebsiella pneumoniae to a fluoroquinolone such as ciprofloxacin (182) has been shown to increase the amount of biofilm by more than 100-fold. Similarly, exposure of K. pneumoniae as well as many other microorganisms to β-lactam agents results in increased production of biofilm (182). Reduction of biofilm can be seen with other agents such as salicylates (183). The reduction of biofilm appears to occur concomitantly with a decrease in porin proteins (184–186). If these porins are being utilized to pump the biofilm precursors to the cell wall outer surface for final assembly, the two events are probably cause and effect. The decrease in biofilm and porin protein has been shown to result in resistance (185,187,188). This phenomenon has been shown to inhibit the activity of cephalosporins (188), aminoglycosides (183), and carbapenems (186). If biofilm is increased by exposure to β-lactam agents, then antimicrobial agents that have an effect on biofilm should be enhanced by preexposure of the bacteria to β-lactam agents. Indeed, this has been noted in both in vitro (189) and in vivo (190) studies.
Effects of Biofilm on Antimicrobial Action
The establishment of biofilm is an important aspect of cell wall physiology for individual cells and is equally important for microorganisms collectively. Bacteria that live and metabolize in these dense biofilm-encased microcolonies gain a number of the advantages enjoyed by multicellular life forms (52,56,57,191). One such advantage is a circulatory system (although primitive) with which to receive nutrients and into which to discharge metabolic wastes. This circulatory system consists of permeable channels that pass through less dense areas of biofilm interspersed within the dense microcolonies (56,191). Along these channels live river populations of microorganisms. These channels have convective flow patterns that permit the penetration and distribution into the biofilm matrix of large (2,000 Da) molecules. Dissolved oxygen is another critical commodity that is distributed within the biofilm through these channels. Microelectrodes have determined that the concentrations of dissolved oxygen in dense microcolonies approach zero at the center, owing to diffusion limitations and oxygen utilization (192). Such direct observations explain the need for anaerobic pathways for microorganisms such as P. aeruginosa (90) and M. tuberculosis (193) that are considered to be strict aerobes. Similar redox-sensitive chemical probes and autoradiography (194) have been used to detect metabolic activity and have demonstrated that, within a microcolony, the majority of cells are metabolically active. Although metabolically active, bacteria within biofilm colonies grow very slowly and are considered to resemble stationary-phase cultures (19,60). Moreover, those microorganisms downriver receive fewer substrates and may therefore become nutritionally starved, setting into motion the complex set of events (59,61) previously described.
Changes in microbial growth rate and nutrient limitation have long been recognized to cause changes in cell envelope components, which, in turn, influence the susceptibility of the microbes to antimicrobial agents (27,59,61,195). Establishment of biofilm is a growth-related factor that influences the susceptibility of the microbes to antimicrobial agents (196). For example, exposure of planktonic cells of P. aeruginosa to a biofilm surface produced by cells of the same species triggers the expression of at least two genes, algC and algG (74). This influences the susceptibility of these cells to antimicrobial agents, because sessile cells encased in biofilm are phenotypically different from planktonic cells of the same species (57,58,197).
The presence of biofilm at the individual cellular level contributes to changes in the overall susceptibility patterns of microorganisms involved in chronic infections, because the encasement by biofilm allows aggregates of cells to exist together in microcolonies. In mature biofilms, bacterial cells occupy only 5% to 35% of the biofilm mass (49,58,63,64,197). There are currently two leading hypotheses for the persistence of chronic biofilm-associated infections: (a) decreased concentrations of antibiotics caused by impaired transport to some regions of the biofilm (49,191,198,199) or by a dilutional effect (49) and (b) physiologic differences of sessile cells (197). A biofilm accumulation model has predicted that both mechanisms would result in reduced antimicrobial susceptibilities of 7-day-old biofilms compared with those of 2-day-old films (49). Growth rate–dependent killing was predicted to be decreased in thicker biofilms because of oxygen depletion, leading to reduced growth rates. The model also predicted resistance to the antibiotic due to depletion caused by increased biomass. The explanation was not that the antibiotic would fail to penetrate the biofilm but instead that the drug would be diluted in the bulk fluid. The binding of agents to biofilm is related to two factors: the relative availability of drug and the relative proximity. Relative availability is proportional to the amount of drug, whereas relative proximity is proportional to the concentration of the drug. The total amount of drug may remain the same as the biomass of polysaccharide increases, but the relative proximity decreases. Finally, many of the factors affecting antimicrobial susceptibility may change over time as the biofilm colony matures, because maturation alters the milieu for many microorganisms within the microcolony. For example, colonies deep within thick, mature biofilm may have reached the starvation stage.
Mechanisms of Antimicrobial Uptake
A factor that is clearly of great importance for effective antimicrobial action against bacteria is the penetration of the antimicrobial agent into both the human cell (200) and the microbial cell (201). Entry of the antimicrobial agents into human cells is considered a part of the pharmacokinetics of these agents (6). In this section, the entry of the antimicrobial agent into the microbial cell will be reviewed. In order to understand antimicrobial uptake, it is useful to understand the physiology of transport mechanisms located on microbial cell membranes.
In all microorganisms, the cytoplasmic membrane provides an osmotic barrier that is permeable to very few substances. It is porous to water and to uncharged organic molecules up to the size of glycerol. Gram-negative bacteria have an additional cell membrane, the outer membrane, which also acts as an effective barrier against antibiotics (155–157). In particular, hydrophobic antibiotics diffuse relatively poorly through the outer cell membrane in gram-negative bacteria in comparison with diffusion through the cytoplasmic membrane. This is due to the lack of glycerophospholipid in the outer portion of the cell bilayer, which instead consists largely of lipopolysaccharides.
Antimicrobial agents derived from microorganisms bear little resemblance to natural substrates brought into the bacterial cell but instead are more akin to metabolites excreted by cells (202). Therefore, with few exceptions (e.g., fosfomycin, which uses a stereospecific nutrient transport system) (203), antibiotics do not utilize active transport mechanisms for substrate uptake into bacteria.
There are three general mechanisms for substrate uptake into the bacterial cell: simple diffusion, facilitated diffusion, and active transport (158,204). There is a fourth mechanism known as the self-promoted uptake pathway, which is used by certain bacteria for the uptake of polycationic antibiotics (205). Each is discussed.
Simple or passive diffusion is defined as movement of molecules across a permeable membrane in which the flux in either direction is proportional to the concentration on the entering side and the rate of net transfer is proportional to the concentration differences between the two sides. An important factor in this type of diffusion is the partitioning coefficient, which essentially indicates the ability of the substrate to dissolve into the membrane interior (i.e., permeability). Simple diffusion kinetics occurs with nonpolar organic molecules such as tetracycline, which penetrates by dissolving in the lipid of the membrane, as well as with antimicrobial agents that move across a membrane through water-filled membrane-protein channel (i.e., pore) that is known as a porin (201,205–208). Fluoroquinolones, for example, are taken into bacteria by passive diffusion through porins in a passive diffusion process that exhibits nonsaturable kinetics. Fluoroquinolones are amphoteric molecules and have both zwitterionic and uncharged forms at neutral pH. Generally, only uncharged molecules are involved in the passive diffusion process, with the amount of uncharged forms greatly influencing the penetrating ability of these agents. Charged molecules can also exhibit passive diffusion, provided there is an electrical gradient across the membrane (209).
Facilitated diffusion in theory involves a barrier-insoluble substance reacting with a carrier (i.e., transporter) within the barrier to form a complex that can shuttle across the membrane, where the substance is then released. This type of diffusion exhibits saturable Michaelis-Menten kinetics with a Km and a Vmax, but the Km is the same on both sides of the membrane because this mechanism is not linked to an energy source. Another name for this type of uptake pathway is a passive carrier–mediated system. Facilitated diffusion is seen in yeasts (210) but has not yet been identified in bacteria.
Active transport means that the bacterial cell has the ability to concentrate molecules within the cell. This ability can be turned on or off (i.e., is inducible) and is linked to an energy source. Without this energy source, the molecules cannot pass across the membrane. The kinetics of active transport exhibits a Km and a Vmax, like the activity of an enzyme, and the carrier system can be saturated.
The self-promoted uptake pathway has been described for gram-negative bacteria and involves binding of the antibiotic to the lipopolysaccharide in the outer membrane (205,211,212). This is followed by outer displacement, by the antimicrobial agent, of magnesium and possibly calcium ions in the lipopolysaccharide matrix of the biofilm (213). This causes instability of the biofilm matrix, as described earlier, and leads to increased permeability (214). The presence of additional divalent cations prevents this by stabilizing the complex. The self-promoted pathway was first described as an uptake mechanism in P. aeruginosa for polycationic antibiotics such as polymyxin and the aminoglycosides (213). More recently, it has been noted for azithromycin in E. coli (215).
Cell membranes of microorganisms must be energized in order to concentrate nutrients needed for growth. The electrochemical gradient–induced proton motive force is a key factor in these energized cytoplasmic membranes. Microbial cell membranes are intrinsically impermeable to protons yet must move protons in or out of the cell. For example, any change in the intracellular pH of the microorganism would need to be absorbed by the buffering capacity of the cytoplasm (148) unless there was some method for expelling protons. Such a pH-homeostatic method exists and involves membrane-bound proton pumps (149). Proton-driven translocation of molecules is facilitated by reduced pH (216). These pumps may at times be responsible for the efflux of antibiotics by pumping out protons that are complexed with a negatively charged antibiotic.
The outer membrane in gram-negative bacteria, through changes in porin diffusion channels, can serve as a permeation barrier and thus greatly influences antimicrobial resistance (156,157,178,217,218). Moreover, the uptake of antimicrobial agents into bacterial cells also can be influenced by efflux mechanisms that may concomitantly act to remove the agent (150,156,219–222). In fact, the antimicrobial activity may be determined by the race between uptake and efflux. It is useful to appreciate the mechanisms of efflux, because these should themselves prove to be excellent targets for antimicrobial agents (222–225).
Membrane-bound proton pumps may be readily overcome by compounds with uncoupling activity (219). Classic uncouplers include carbonyl cyanide-m-chlorophenylhydrazine and 2,4-dinitrophenol (226). These uncouplers result in the abolition of respiratory control in the bacteria. This, in turn, results in stimulation of respiratory activity.
INHIBITORY OR LETHAL ANTIMICROBIAL ACTIVITY VERSUS FUNCTIONAL SYNERGY AGAINST MICROBES
Inhibitory and Lethal Effects
The goal of antimicrobial therapy, as appreciated by Lister and Ehrlich, is to destroy the invading microorganism without harming the host. The effectiveness of an antimicrobial agent has traditionally been measured by its ability to inhibit and kill bacteria. In theory, there are three basic ways to kill a bacterial cell: by causing irreparable damage to its genome, to its envelope, and to certain classes of its proteins (114–121). Antimicrobial agents have been developed that attempt to kill bacteria in each of these ways. Often, several antibiotics that use two of these three different ways are combined to enhance the lethal effect. Yet, as already noted, bacteria are not particularly easy to kill. This fact has not escaped microbiologists. It is well known that most antimicrobial agents exert their lethal effects on bacteria during the growth phase (2,27,195). Therefore, microbiologists have designed routine susceptibility tests to measure antimicrobial activity during the logarithmic growth phase in media that provide all of the ingredients for optimal growth (62). However, this is not the usual state of microorganisms in infected tissues. For example, S. aureus isolates from tissue-cage infections in rats have been shown to be in a state of dormancy and thus are relatively resistant to most antimicrobial agents (227). Perhaps the closest that broth susceptibility testing comes to mimicking a clinical infection is in the case of acute bacterial meningitis. Even then, there clearly is room for improvement (228). Susceptibility testing must be repositioned to provide test results that correlate with the clinical infection. Fortunately, clinical microbiologists have been working to accomplish this goal. An example is the integrated use of pharmacokinetic and pharmacodynamic models for the definition of breakpoints (9). As a result, the correlation of in vitro susceptibility testing with in vivo clinical effectiveness has markedly improved (14). Moreover, clinical microbiology laboratories are aware of emerging mechanisms of resistance and are particularly vigilant in detecting such resistance (229,230). Finally, antimicrobial susceptibility testing is frequently integrated with an antimicrobial stewardship program (231) that is aimed at controlling resistance.
Functional Synergy
Most clinical infections involve bacteria in a sessile state, as opposed to a planktonic state (63). However, antimicrobial agents that are able to kill bacteria in their sessile state are few in number (28,232,233). Of those agents currently available for clinical use, carbapenems and the fluoroquinolones have the greatest lethal effect against sessile bacteria—a lethal effect more readily obtained against gram-negative isolates than against gram-positive ones (233–235). Against gram-positive pathogens such as staphylococci, daptomycin has the greatest lethal effect against stationary-phase and nondividing organisms (236,237). This in vitro bactericidal activity, often defined as equal to or greater than a 3 log10 decrease in colony-forming units over a 24-hour period (62), may not be sufficient for total microbial killing against certain microorganisms (48) or with certain infections such as endocarditis (238). Total and rapid microbial killing is important in acute bacterial meningitis as well as in acute sepsis in immunodeficient hosts (2,7). Even the most rigorous in vitro susceptibility test methods, including time-kill kinetic methodology, may provide misinformation if the growth phase of the microorganism and the clinically desired end point are not correlated with the test method (14). This is shown by a report by investigators who found that clarithromycin, like other macrolides, demonstrated in vitro bactericidal activity against pneumococci by time-kill kinetic methodology (239). However, in a rabbit model for pneumococcal meningitis, clarithromycin was unable to cure pneumococcal meningitis despite susceptible isolates and cerebrospinal fluid levels of clarithromycin comparable to those used to achieve in vitro killing (239). In chronic infections such as endocarditis and osteomyelitis, where involvement of biofilm is almost always present, rapid microbial killing is usually not achievable due to factors such as decreased biofilmpenetration/dilutional effects and dormant growth phase (240,241). Total microbial killing, on the other hand, is a well-recognized goal when treating these particular infections; antimicrobial therapy is usually given for 6 to 8 weeks in order to achieve total microbial killing (242,243).
The use of bactericidal drugs in order to achieve total microbial killing is not required for most infections (244,245). However, when bacterial eradication is deemed necessary, there are a number of ways that this may be predicted (246). Pharmacokinetic and pharmacodynamic parameters may be useful for predicting bacterial response (246,247). In addition, there are bactericidal tests including proposed reference methods available in most clinical microbiology laboratories (248).
The accumulated knowledge of antimicrobial mechanisms of action on different growth phases of bacteria has reached a point where it may encourage a multicomponent drug approach to antimicrobial therapy (21,249,250). This approach may involve using combination therapy that achieves functional synergy against the infecting microorganism. Functional synergy directed against the microorganism often can be achieved by using knowledge of the various physiologic states within which microorganisms exist combined with knowledge of specific antimicrobial agents that interfere with each of the physiologic states. When these agents are combined, their use may result in enhanced microbial killing that can be thought of as functional synergy. Although enhanced killing, as defined by a strict definition of synergy, may not be detected using in vitro methods, enhancement of total killing may be measured in other ways. For example, animal models have long been used to assess the ability of antimicrobial agents alone or in combination to eradicate microorganisms (251).
Functional synergy as a therapeutic approach is already in use but is not well appreciated. The therapy of TB is one of the oldest examples of this approach, for in such therapy, multiple antituberculous agents result in enhanced mycobacterial killing as well as minimizing the emergence of resistance. An example of this therapeutic approach is the recognition of the lethal effect of metronidazole on dormant forms of M. tuberculosis (252). This lethal activity appears to be related to the fact that the dormant state requires anaerobic pathways, which then provide the necessary electrons to activate metronidazole to its electrophilic degradation products (252). Metronidazole alone does not reduce the bacillary burden of M. tuberculosis in the guinea pig model (253). Exposing M. tuberculosis in its dormant state to metronidazole apparently kills the organism or triggers aerobic respiration. The return to aerobic pathways does not occur independently but instead occurs with resumption of mycobacterial replication. Thus, combining metronidazole or similar agents with agents that interfere with the replicating stage creates functional synergy that allows enhanced killing of the mycobacterium (24,254). Drug development strategies that target the different physiologic states of M. tuberculosis including the latent phase may allow improved treatment of TB (255). Indeed, the addition of metronidazole to isoniazid (INH) and rifampin in the macaque model has been shown to effectively treat animals with active TB within 2 months (256).
Another example of functional synergy is the use of an aminoglycoside with a β-lactam agent for the therapy of infections such as bacterial endocarditis caused by P. aeruginosa. The increased effectiveness of this combination is due to the disruption of the biofilm by the aminoglycoside (173,257), which enhances the β-lactam agent in two ways. The first is when disruption provides holes in the bacterial cell wall that allow increased penetration of the β-lactam agent. The second is when the dormant form is forced to replicate in an attempt to fix the damaged cell wall, thus providing a target for the β-lactam agent.
Other chronic infections that have benefited from a multidrug approach that provides functional synergy include pulmonary infections caused by P. aeruginosa in patients with cystic fibrosis. These infections involve the establishment in lung tissue of biofilm-encased microcolonies in which are found some dwarf forms, which may represent dormant forms that are utilizing anaerobic pathways (90,104,105). Clinical cure of these Pseudomonas pulmonary infections is rarely achieved (104). This is consistent with the in vitro observation that total microbial killing of sessile strains of P. aeruginosa is extremely difficult to achieve after the biofilm has matured for 5 to 7 days (196). Neither older synergistic combinations such as tobramycin and piperacillin (77) nor newer synergistic combinations such as fosfomycin and ofloxacin (258) are able to achieve total killing.
There are, however, some approaches that may allow functional synergy. The use of aerosolized tobramycin (259,260) or aerosolized colistin (261) is one of these; this approach provides greater concentrations of a biofilm disrupter (i.e., both tobramycin and colistin) as well as a bacterial cell membrane disrupter (i.e., colistin), which then can enhance the systemic use of other antipseudomonal agents. The prolonged use of aminoglycosides in chronic Pseudomonas infections is known to be followed by the emergence of aminoglycoside-resistant Pseudomonas strains characterized by a deep rough colony morphology on agar plates due to the lack of biofilm (257). The lack of a lipopolysaccharide/biofilm target for the primary action of the aminoglycoside is the mechanism of resistance, because these strains do not exhibit altered ribosomes or produce aminoglycoside-inactivating enzymes. The outer cell walls of these aminoglycoside-resistant strains are characterized by the lack of lipopolysaccharide and a marked increase in the amount of OprH OMP (257). Overproduction of this outer cell membrane protein decreases the accumulation of polymyxin and gentamicin by the self-promoted pathway (214). However, overproduction of OprH is associated with increased susceptibility to fluoroquinolone antibiotics (262). It appears that the overproduction of OprH is a mechanism that minimizes biofilm in order to counter the effects of biofilm disruption by polycationic agents, but in doing so, the altered cell wall seems to offer increased diffusion of lipophilic fluoroquinolones into the cytoplasm. The use of a fluoroquinolone and an antipseudomonal β-lactam agent along with the aerosolized aminoglycoside (264) thus allows functional synergy and provides an additional therapeutic option (265). Moreover, the addition of a macrolide such as azithromycin or clarithromycin to this regimen may prove useful. These macrolides have been shown to decrease the production of both biofilm (135,137,266) and exoenzymes (131,132) by P. aeruginosa, which creates yet another biofilm-related conflict while preventing further pulmonary damage by the exoenzymes (132,266–271). Macrolides and ketolides have been shown to have an effect on the outer membrane of P. aeruginosa (272,273) that appears to potentiate the activity of antipseudomonal agents and may allow functional synergy (274). Clinical experience with the use of aerosolized tobramycin (259,260), fluoroquinolones (264), and macrolides (275) for exacerbations of Pseudomonas infections in cystic fibrosis patients have shown that multidrug regimens that include one or more of these three agents result in reductions in P. aeruginosa sputum density and improvements in pulmonary function (265). Interestingly, a similar approach using a combination of clarithromycin and ceftazidime in a rat model for foreign body–related osteomyelitis caused by P. aeruginosa demonstrated that clarithromycin eradicated the biofilm and enhanced the bactericidal effect of ceftazidime (276).
There are other examples of functional synergy in microbes that can be purposely created by the selective use of antibiotics. Enhanced microbial killing is a frequent goal of combination antimicrobial therapy for infective endocarditis (20). Therefore, a number of examples of functional synergy demonstrated in experimental endocarditis are discussed. Temafloxacin has been shown to be effective in the therapy of experimental streptococcal endocarditis, and studies have found it to penetrate vegetations in a homogeneous manner (277). Dextranase is an enzyme capable of hydrolyzing 20% to 90% of streptococcal glycocalyx (biofilm). When used alone, dextranase has no in vitro antimicrobial effect on viridans streptococci nor does it have a beneficial effect on experimental streptococcal endocarditis (278). When dextranase is used in combination with temafloxacin, it significantly potentiates the effect of temafloxacin in vivo by reducing the amount of bacterial biofilm in infected vegetations and by altering the metabolic status of the microorganisms (279). The same effect has been seen when dextranase and penicillin have been combined in the treatment of experimental streptococcal endocarditis (278). Finally, an animal model for experimental P. aeruginosa endocarditis has shown an identical effect for alginase combined with amikacin (280). Of importance is the lack of beneficial effect demonstrated when the vegetation size is reduced by fibrinolytic therapy alone (281,282). The results of these studies are consistent with the theory that the bacteria in microcolonies embedded in biofilm have a lower metabolic rate (283). Reducing the amount of biofilm results in both an increased metabolic rate and increased replication, which each increases the antimicrobial activity of most antibiotics.
Another experimental approach to disrupting the biofilm as a method for creating functional synergy is to use the proteolytic enzyme serratiopeptidase (284). Serratiopeptidase is a metalloprotease produced by a strain of Serratia that is only partially inhibited by in vivo protease inhibitors and has been used as an antiinflammatory drug because of its ability to increase the penetration of antibiotics into infected sites (285). This protease has been found to enhance the activity of ofloxacin on sessile cultures of P. aeruginosa and S. aureus (286). In addition, serratiopeptidase has been shown to reduce the ability of Listeria monocytogenes to form biofilm and to invade host cells (287).
Another commonly used drug with the potential for creating biofilm-related functional synergy in the therapy of microbial infections is aspirin (288). Aspirin has been noted to be a cell wall permeabilizer for P. aeruginosa (178). This role as a cell wall permeabilizer may be related to its effect on biofilm. Aspirin has been shown to cause a dose-dependent reduction in the weight of aortic vegetations in experimental endocarditis (289). In addition, when combined with vancomycin, aspirin improves the sterilization rate of aortic valve vegetations infected with S. aureus. These effects are similar to those of dextranase (279) and the protease of Serratia (286) and may be a result of functional synergy. Aspirin has been found to diminish the amount of microbial biofilm in a number of other studies (183,288). Salicylates also are known to depress the synthesis of porins in E. coli, K. pneumoniae, Serratia marcescens, Burkholderia cepacia, and P. aeruginosa (184–186,188). If these depressed porins are involved in the efflux of biofilm precursors as a part of biofilm maintenance, these two physiologic phenomena may be related. Finally, the combination of aspirin and amphotericin B has demonstrated functional synergy against biofilm cells of Candida albicans and Candida parapsilosis versus indifferent effects against planktonic cells of these microorganisms (290). Each of these antimicrobial strategies against infectious bacterial biofilm (291) is an example of the use of functional synergy.
Finally, rifampin combination therapy is a very controversial multidrug approach to a number of nonmycobacterial infections (292,293) that may owe its somewhat surprising albeit unpredictable efficacy to functional synergy. Rifampin combination therapy has been used clinically for various types of infections (293), but the predominant use seems to be for staphylococcal infections (292) that involve osteomyelitis and/or a prosthetic device–related infection. The controversy stems from a lack of convincing in vitro data that support the in vivo clinical findings. There are a number of clinical studies involving bone or joint infections that have demonstrated such in vivo efficacy, although these studies are generally underpowered (294–301). Examples of a number of these clinical studies are provided. In one study (296), therapy with 900 mg/day rifampin plus 600 mg/day ofloxacin for 6 months was used for patients with prosthetic implants infected with Staphylococcus spp. The overall success rate was 74% among 47 patients, with 62% of patients being cured without removal of their orthopedic device (296). The success rate was 81% for the hip prosthesis group, 69% for the knee prosthesis group, and 69% for the osteosynthesis device group. A total of eight treatment failures were related to the isolation of a resistant microorganism. In another study (297), 33 patients with culture-proven staphylococcal infection associated with stable orthopedic implants and with a short duration of symptoms of infection were treated: 18 patients received ciprofloxacin and rifampin, whereas 15 patients received ciprofloxacin and a placebo. Twenty-four patients completed the trial; the cure rate was 12 of 12 (100%) for those who received ciprofloxacin plus rifampin versus 7 of 12 (58%) for those who received ciprofloxacin plus a placebo (297). In a third study (295), 10 patients with Staphylococcus spp–infected orthopedic implants were treated with various antibiotic regimens, all of which included rifampin. Of these patients, 8 were cured. Many of these studies (296–298,301) combined rifampin with a quinolone; indeed, early results with such oral therapy using rifampin combined with a quinolone have been encouraging (302).
Animal models of adjunctive rifampin for therapy of Staphylococcus-infected prosthetic devices/foreign bodies/osteomyelitis may offer some additional insight. A rat model of chronic staphylococcal foreign body infection (227) demonstrated that antimicrobial combinations of fleroxacin plus vancomycin and vancomycin plus fleroxacin and rifampin were highly effective and superior to single drugs. Further, the three-drug therapeutic regimen decreased bacterial counts more rapidly than the two-drug therapy and was curative in most cases (92% for three drugs versus 41% for two and less than 6% for monotherapy). No mutants resistant to these three agents were detected with combination therapy. A rabbit model of acute staphylococcal osteomyelitis caused by MRSA noted a 100% infection clearance with tigecycline and rifampin versus a 90% clearance with tigecycline alone, whereas vancomycin and rifampin showed a 90% infection clearance versus an 81.8% clearance with vancomycin alone (303). A guinea pig model assessing linezolid alone or combined with rifampin against a foreign body infection caused by MRSA demonstrated that the efficacy in the eradication of cage-associated MRSA infection was achieved only with the combination of rifampin and linezolid, with cure rates being between 50% and 60%; in this guinea pig model, a levofloxacin–rifampin combination achieved a 91% cure rate against a quinolone-susceptible MRSA strain (304). An MRSA knee prosthesis infection in rabbits was used to evaluate daptomycin or vancomycin alone and in combination with rifampin (305). This study demonstrated that daptomycin combined with rifampin sterilized 11 of 11 bones versus 2 of 12 for daptomycin alone, whereas vancomycin combined with rifampin sterilized 6 of 8 bones versus 0 of 12 for vancomycin alone (305). Moreover, rifampin prevented the emergence of daptomycin-resistant MRSA; the authors concluded that adjunctive rifampin is crucial to optimizing daptomycin efficacy against rabbit prosthetic joint infection due to MRSA (305). Additional studies in animal models have demonstrated similar efficacy of rifampin combinations (306–308).
The success of adjunctive rifampin for the therapy of Staphylococcus-infected prosthetic devices/foreign bodies/osteomyelitis may once again be due to functional synergy that targets biofilm (241,291). The efficacy of fluoroquinolones (302) is of interest because fluoroquinolones have been shown to decrease the production of slime (biofilm) by S. epidermidis (181), and it is likely that rifampin, through inhibition of protein synthesis, may decrease or prevent the availability of critical enzymes needed for ongoing maintenance of biofilm. As the biofilm microcolonies attached to the prosthetic device or glued into the bone begin to be slowly disrupted by the lack of ongoing maintenance, the staphylococci are forced to replicate, which further enhances the antimicrobial action of each antibiotic.
MECHANISMS OF ACTION FOR SELECTED CLASSES OF ANTIMICROBIAL AGENTS
Antimicrobial Classes in Current Clinical Use
β-Lactam Agents
As previously described, the main structural features of the peptidoglycan sacculus are linear glycan chains interlinked by short peptide bridges. A number of enzymatic activities are involved in the biosynthesis of the sacculus: catalyzation by glycosyltransferase enzymes of the formation of the linear glycan chains, cross-linking of the glycan chains by transglycosylase enzymes, and cross-linking via peptide bridges by transpeptidase enzymes (32,151). The latter peptide cross-links provide mechanical strength against osmotic pressure forces. Peptidoglycan structural modifications of completed cell wall are required in replicating cells as they grow, and each microorganism therefore possesses specific peptidoglycan hydrolases that are responsible for such structural adjustments (38–40,309). It is these transpeptidases/hydrolases (39,40,309), as well as other factors such as activation of newly recognized apoptotic death pathways (42–45), that appear to be important target(s) of β-lactam agents (36,40,96,310–314).
The mechanism of action of β-lactam agents is more complex than initially thought and likely involves three interrelated cellular processes (314). The first of these cellular processes is transpeptidation, which initially was thought to be the sole target of β-lactam agents (310). Penicillins, because of their structural similarity to the C-terminal D-alanyl-D-alanine end of the peptide stem, react chemically with the transpeptidases, also known as PBPs, to form stable acyl-enzymes, inactivating the PBPs and preventing further cross-linking. The inhibition of glycan cross-linking then leads to a weakened cell wall, which eventually ruptures due to osmotic pressure. However, it was noted that penicillins were able to cause inhibition of growth in certain bacteria without bacteriolysis. Therefore, triggering of autolytic cell wall enzymes was considered as a second and separate target of β-lactam agents (43,127,311). However, the mechanism for control of the autolytic system and how it was activated during treatment with β-lactam agents remained unknown until a number of observations suggested several possibilities. The electrophysiologic state of the cellular membrane is thought to be an important factor in the regulation of bacterial cell wall autolysis (107,315,316). There is increasing evidence that β-lactam agents may depolarize the membrane potential as a signal to induce autolysis (44,45,317). Moreover, there are a number of regulatory genes that are involved in bacterial autolysis (97,318,319). These genes may be activated after exposure to a sufficient concentration of β-lactam agent to cause irreparable damage (30,95,319). For example, a signal transduction pathway involved in regulating apoptotic death in pneumococci has been described (96). One of the death signals appears to be a peptide, which may function in a quorum-sensing manner. Finally, metabolism-related depletion of NADH, leaching of iron from iron-sulfur clusters, and stimulation of the Fenton reaction has been shown to lead to formation of harmful hydroxyl radicals that triggers an oxidative damage cellular death pathway (44,45). This pathway may be modulated by the stringent response in a manner yet to be detailed. Modulation of the stringent response under antimicrobial selection appears to create mutants that are virulent and not killed by a broad spectrum of antimicrobial agents (55). This resistance phenomenon has been described as physiologic tolerance (28,320).
Penicillins
Penicillins are characterized by a four-membered β-lactam ring fused to a five-membered thiazolidine ring containing a side chain (321). Manipulations of this side chain have been important in the pharmacokinetics and pharmacodynamics of penicillins. The ability to produce 6-aminopenicillanic acid (6-APA) by fermentation allowed chemists to replace the amino group of 6-APA with a large number of altered side chains, thus producing many different semisynthetic penicillins (322). The steric hindrance around the amide bond produced by bulky side chains such as carbocyclic or heterocyclic rings with substituents at the orthoposition of the 6-APA site produced the first semisynthetic penicillins with increased stability against staphylococcal β-lactamase. A number of such antistaphylococcal penicillins with bulky side chains have been synthesized, including methicillin; nafcillin; and the isoxazolyl penicillins, oxacillin, cloxacillin, dicloxacillin, and flucloxacillin.
Semisynthetic penicillins also include those created by a simple replacement of the α-carbon of the hydrophobic side chain at position 6 of benzylpenicillin by an amino (e.g., ampicillin), a carboxyl (e.g., carbenicillin), a ureido (e.g., mezlocillin), a piperazino (e.g., piperacillin) group, or a methoxy (e.g., temocillin). The result was the development of the extended-spectrum penicillins, which have been grouped as aminopenicillins, carboxypenicillins, ureidopenicillins, and methoxypenicillins (322–325). Such substitutions provided improved penetration through the outer cell membrane of gram-negative microorganisms as well as increased stability against β-lactamases produced by these pathogens. In particular, penetration through the restrictive pores of P. aeruginosa resulted in antipseudomonal activity for the carboxypenicillins (e.g., carbenicillin and ticarcillin) and the ureidopenicillins (e.g., mezlocillin, azlocillin, and piperacillin). The only methoxypenicillin in clinical use is temocillin (324,325), which is not active against gram-positive organisms, anaerobes, and Pseudomonas species; temocillin has not been approved for use in the United States. Temocillin is, however, resistant to most if not all classic and extended-spectrum β-lactamases as well as to AmpC β-lactamases (324). For this reason, it has been used in England as a carbapenem-sparing agent (324,325). Because the stability against β-lactamases did not include staphylococcal β-lactamase, a number of these extended-spectrum penicillins were combined with a β-lactamase inhibitor (e.g., clavulanate, sulbactam, or tazobactam) and are known as β-lactam–β-lactamase inhibitor combinations (323,326,327). To date, these combinations include amoxicillin/clavulanate, ampicillin/sulbactam, ticarcillin/clavulanate, and piperacillin/tazobactam.
Cephalosporins
Cephalosporins are characterized by a four-membered β-lactam ring fused to a sulfur-containing ring-expanded system (328). One of the first cephalosporins, cephalosporin C, possessed an aminoadipic side chain, which could easily be chemically removed to give rise to 7-aminocephalosporonic acid (7-ACA), which is analogous to 6-APA (329). From 7-ACA came the first-generation semisynthetic cephalosporins such as cefazolin. Substitutions at the C7 position as well as at the C3 position of the dihydrothiazine ring allow greater variation of semisynthetic cephalosporins than can be achieved with the penicillins (322,323). Consequently, more cephalosporins have been developed, and detailed reviews of these agents are available (322,323,328,330). Side chain substitution in the cephalosporins is built on the experience with penicillins and includes thiazolyl and phenylglycyl side chains. The substitutions at the C7 position are of particular importance in governing stability against β-lactamases. For example, substitution at the C7-α position of cephalosporins with a methoxy group (e.g., cefoxitin and cefotetan, which are second-generation cephalosporins) resulted in the cephamycins, which have increased stability against β-lactamases, including those of the Bacteroides fragilis group. Substitution at the C7-β position with a methoxyimino group (e.g., cefotaxime, which is a third-generation cephalosporin) also increased the resistance of these agents to β-lactamases. Acyl side chains used with cephalosporins include aminothiazole oximes, which may have charged carboxylates (e.g., ceftazidime, which is a third-generation cephalosporin) that improve penetration through gram-negative bacterial outer membranes. Cefepime, a fourth-generation cephalosporin, also has a positively charged quaternary ammonium in position C3, which creates a zwitterion that allows increased penetration of the gram-negative bacterial outer membrane. The 2-aminothiazolylacetamido group found in cefepime provides increased stability against β-lactamases. Ceftaroline is the first member of a new subclass of β-lactam agents, cephalosporins with activity against MRSA (331). A 1,3-thiazole ring attached to the 3-position of the cephalosporin nucleus and the oxime group in the C7 acyl moiety provide the basis for increased activity against MRSA with the 1,3-thiazole ring binding tightly to the MRSA PBP 2A following a conformational change in the protein allowing the active site to be exposed for binding (332). Ceftaroline is also active against multidrug-resistant S. pneumoniae.
Carbapenems
Carbapenems differ from conventional penicillins in having no sulfur atom in their five-membered ring and in having a double bond between carbons 2 and 3 (332,333). This sterically alters the cis/trans configuration of the molecule in comparison with other β-lactam agents and places the amide bond away from the water-containing groove of the serine-based β-lactamases. However, carbapenems are susceptible to hydrolysis by metallo-β-lactamases. Four carbapenems (imipenem, meropenem, ertapenem, and doripenem) are approved for clinical use in the United States.
Monobactams
There are two monocyclic β-lactams produced by microorganisms that possess antimicrobial activity, nocardicins and monobactams (334). The monobactam nucleus of these compounds exhibits only weak antimicrobial activity, and they, like the penicillins and cephalosporins, must have substitution around the central nucleus to achieve clinically useful antimicrobial activity. Side chain structure–activity relationships in monobactams parallel those of penicillins and cephalosporins. There is only one monobactam antibiotic, aztreonam, that has a 3-acyl aminothiazole-oxime side chain identical to that of ceftazidime, while the lactam ring has a N-sulfonate substituent on the other side (330,332,334). The N-sulfonate substituent is essential for β-lactamase stability. Like ceftazidime, aztreonam is useful only against gram-negative pathogens, with good activity against P. aeruginosa.
Aminoglycosides
All aminoglycoside antibiotics contain one or more amino sugar residues linked to a central, six-membered aminocyclitol ring by pseudoglycosidic bond(s). Spectinomycin is, strictly speaking, also an aminocyclitol with three fused rings but lacks amino sugars and pseudoglycosidic bonds. The primary mechanism of action of aminoglycosides is a decrease in protein synthesis after the drug has bound to the prokaryotic ribosome at the 16S ribosomal RNA (rRNA) site located in the small (30S) subunit of the ribosome (335–339). Aminoglycosides are hydrophilic sugars with multiple amino groups that function as polycations. Their polycationic nature allows binding to the polyanionic 16S rRNA on the 30S ribosome at the A site for aminoacyl–tRNA binding (338,339). The A site is composed of portions of the 530 loop, helix 34, and the base of helix 44; transfer RNA (tRNA) anticodons bind in a cleft formed between these individual domains of the A site. Binding of the aminoglycoside to the A site inhibits the translation process by causing misreading and/or hindering the translocation step. Analysis by X-ray crystallography suggests that the polycation binds to the RNA bases rather than to backbone atoms. Moreover, attachment at the A site suggests that aminoglycosides block a required transformational transition during the peptide bond–forming translocation process, bringing the translocation steps of protein synthesis in the ribosome to a halt. Aminoglycosides also bind to helix 69 of the large (50S) subunit of the ribosome (340,341). The result of this binding is reduction in the mobility of an adenine residue at position 1492 of the rRNA A-site; this reduction in mobility may be a key determinant in the antibacterial activity of aminoglycosides (342).
The polycationic nature of aminoglycosides also accounts for their recognized effect on biofilm and cell membranes (163,172). Aminoglycosides are bactericidal agents and often exhibit a rapid lethal effect on susceptible aerobic gram-negative bacilli. Such a rapid lethal effect has been noted to be contrary to the expected effect of agents acting on ribosomal targets (173). This lethal effect of aminoglycosides against aerobic gram-negative bacilli, moreover, is concentration-dependent, with increasing concentrations achieving increased killing. Their effect against gram-positive cocci is, at best, inhibitory, unless a β-lactam agent is used in combination with the aminoglycoside.
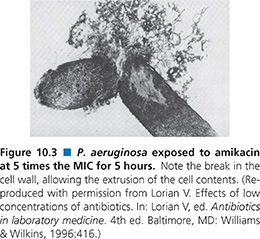
Inhibition of protein synthesis, however, usually does not produce a bactericidal effect, let alone a rapid one. Therefore, binding to the 30S ribosome may not be the only mechanism of antimicrobial action for aminoglycosides; in fact, many susceptible gram-negative bacilli may be dead long before the drug arrives at the 30S ribosome (173,174,212). It is now recognized that aminoglycosides are polycations that competitively displace cell biofilm–associated Mg2+ and Ca2+ linking the polysaccharides of adjacent lipopolysaccharide molecules (174,211–213). The result is shedding of cell membrane blebs, with formation of transient holes in the cell wall and disruption of the normal permeability of the cell wall (163,170,173,174). This action alone may be sufficient to kill many susceptible gram-negative bacteria before the aminoglycoside has a chance to reach the 30S ribosome (Fig. 10.3). The surface effect of gentamicin has been investigated using bovine serum albumin–gentamicin complexes (174), which have been shown to be bactericidal against P. aeruginosa. These findings are in agreement with similar studies done with other immobilized surface agents (146,343–345).
Increased understanding of the mechanisms of action of aminoglycosides brought with it the realization that aminoglycosides have a concentration-dependent bactericidal effect (346) as well as a considerable postantibiotic effect (2). This allowed the dosing schedule to be modified to once per day (347). The modification integrated both pharmacokinetic and pharmacodynamic properties and offered the potential for greater efficacy and less toxicity (8,347–350). There has now been considerable experience with once-daily aminoglycoside regimens (347–350). Such regimens indeed appear to be clinically effective while reducing the incidence of nephrotoxicity. Moreover, they reduce the cost by decreasing ancillary service time and the need for serum aminoglycoside determinations.
Another question about the optimal utilization of aminoglycosides can be addressed. This concerns the timing of the doses when both an aminoglycoside and a β-lactam agent are administered. In vitro (189) and in vivo (190) studies have clearly demonstrated a remarkable advantage gained from nonsimultaneous administration of aminoglycosides and β-lactam agents when they are used in combination. The main benefit is a marked delay in bacterial regrowth regardless of the order in which the agents are given. However, the initial bactericidal effect is greater when the aminoglycoside is given first. The minimum time interval between doses of the aminoglycoside and the β-lactam agent for maximum delay of regrowth is 2 hours.
The reason for this phenomenon is clear if one accepts the premise that the aminoglycoside has its primary antimicrobial effect on biofilm. This effect would be greatest when there was no prior or concomitant use of cell wall–active agents, which have been shown to markedly affect the bacterial cell surfaces (103) and would thus lessen the opportunity for the aminoglycoside to have the optimal biofilm targets.
The bactericidal effects of aminoglycosides combined with β-lactam agents in vitro have been shown to be dependent on the concentrations of β-lactam agents used. If the concentration of the β-lactam agent is not optimized for bactericidal activity (i.e., four- to eightfold higher than the MIC), the effect of the β-lactam on the test microorganism is to stimulate increased production of biofilm (182) and thus enhance the effect of the aminoglycoside by providing a better target. Lorian and Ernst (351) have demonstrated this concept nicely. If, on the other hand, the concentration of the β-lactam agent is high enough to disrupt the cell wall morphology (103), then the biofilm target is lessened, which in turn lessens the lethal effect of the aminoglycoside (174,196). These two effects are exactly the opposite of what would be predicted to occur if the target of the aminoglycoside was the ribosome rather than biofilm. The overall lethal effect of aminoglycosides in vivo is enhanced by administering the aminoglycoside before the β-lactam agent (190). Historically, aminoglycosides have been used in combination with other antimicrobial agents (most often a β-lactam agent) in order to enhance microbial killing and achieve increased efficacy (349). Most studies, however, fail to demonstrate improved outcomes (349); this may be due, in part, to not administering the aminoglycoside first.
Macrolides, Azolides, Lincosamides, Ketolides, and Streptogramins
The macrolides, azolides, lincosamides, ketolides, and streptogramins are grouped together despite structural differences because of similar biologic properties, including their mechanism of action against the 50S subunit of the bacterial ribosomes. Bacterial ribosomes are an important target for antimicrobial agents and have two specific subunits that are targeted, the 30S ribosomal subunit and the 50S ribosomal subunit (118). The specific target for the 50S ribosomal subunit for many of these agents appears to be domain V of the 23S rRNA, which is the peptidyltransferase center (352,353). Macrolides block the approach to the exit tunnel for elongating peptides and thus prevent polypeptide translation, causing premature release of peptidyl–tRNA intermediates. Lincosamides, however, inhibit the initiation of peptide chain formation (134,354), whereas the effect of the other macrolides is to prevent the extension of the growing peptide chain. Macrolides also block assembly of 50S subunits by their interaction with the 23S rRNA. There are interspecies variations in the ribosome structure that affect the binding of macrolides (355). These interspecies variations not only affect wild-type ribosomes but also affect ribosomes that have acquired resistance mutations. Thus, a mutation that results in high-level resistance to a specific macrolide in a particular species may only produce low-level resistance or none at all in a different species (355,356). Finally, it should be noted that the 23S rRNA of bacteria is the homolog of human mitochondrial 16S rRNA; antimicrobial agents that inhibit the bacterial 23S peptidyltransferase center may also impair human mitochondrial function (357).
Ketolides are the latest members of the macrolide group and are novel semisynthetic 14-membered–ring macrolides in which the main structural innovations are the lack of the neutral sugar cladinose in position C3 as well as a C11/C12 carbamate (358–360). When the C3 cladinose sugar moiety is removed, the resulting 3-hydroxy group is oxidized to a 3-keto group, hence the name ketolide. The macrolides, azolides, lincosamides, and ketolides all appear to bind to the same site or contiguous sites on the ribosome and so they may become competitive inhibitors if used together (359,361,362). This mechanism of action, inhibition of protein synthesis, results in bacteriostatic activity against most bacteria by all of these agents except ketolides, which have bactericidal activity (358–360). Inhibition of critical proteins in certain microbial species does, however, result in bactericidal activity of the macrolides. Such species-specific bactericidal activity is seen in vitro with the macrolide clarithromycin against S. pneumoniae but is less evident in vivo (239).
Macrolide antibiotics such as erythromycin, clarithromycin, and azithromycin appear to have the ability to decrease sputum production in patients with chronic respiratory infections (270,271,362–364). This has been attributed to a direct effect on the production of sputum (363) but may instead be due to inhibition of biofilm production by respiratory pathogens (266,270). Macrolides have been shown to markedly reduce the biofilm structure of microorganisms in their sessile phase (135) and to reduce the amount of virulent exotoxins (131,132). These microorganisms include S. epidermidis and P. aeruginosa. The mechanism seems to involve the suppression of a step or steps in the synthesis of monosaccharides, probably as a result of the suppression of mRNA (130,138). The use of macrolides such as erythromycin or azithromycin has been beneficial in chronic respiratory tract infections caused by P. aeruginosa (268–271).
Erythromycin. Erythromycin is a metabolic product of Streptomyces erythreus and consists of a 14-member lactone ring to which are attached two deoxy-sugars, desosamine and cladinose. The macrocyclic lactone ring is the source of the class name, macrolide. Erythromycin, like most macrolides, appears to act by binding in the ribosomal tunnel through which the nascent peptide moves and thus can be considered a peptidyltransferase inhibitor (130). Against some rapidly replicating bacteria, erythromycin exhibits in vitro bactericidal activity, but overall, it is considered to be bacteriostatic in clinical use.
Clarithromycin. Clarithromycin, like erythromycin, has a 14-member lactone ring structure that has been altered by the addition of a methoxy group at C6 of the lactone ring (133,361,362). This substitution primarily results in better oral absorption, with little effect on the spectrum of activity. In fact, erythromycin, clarithromycin, and azithromycin appear to bind to the same receptor on the bacterial 50S ribosome subunit. Clarithromycin, like the other macrolides, has species-specific bactericidal activity (239), as defined by in vitro methods of assessment where a greater than or equal to 3 log10 decrease in colony-forming units over a 24-hour period is defined as bactericidal (62). The species usually considered to be killed by macrolides are S. pneumoniae, S. pyogenes, and H. influenzae. However, the bactericidal activity of clarithromycin against susceptible strains of S. pneumoniae, as demonstrated in vitro by time-kill kinetic curves, has been found to lack correlation with results from the rabbit model for pneumococcal meningitis (239). The use of an in vitro method to measure total microbial killing has been suggested as a way to better assess the bactericidal activity of antimicrobial agents used in bacterial meningitis (228).
Azithromycin. Azithromycin is derived from erythromycin and differs in having a methyl-substituted nitrogen in its 15-membered lactone ring (362,365). This class of drugs receives its name, azolides, from the presence of the nitrogen group. Azithromycin has the same mechanism of action as erythromycin, and these two drugs bind so close to each other on the ribosome that they are considered competitive inhibitors. Azithromycin, like erythromycin and other macrolides, is a bacteriostatic agent. Azithromycin, however, has a major advantage, namely, its absorption and prolonged intracellular/interstitial fluid levels. In particular, the intracellular levels should greatly enhance the therapy of infections caused by intracellular pathogens.
Dirithromycin. Dirithromycin is a semisynthetic derivative of erythromycin that is converted during absorption and distribution to an active metabolite 9-(S)-erythromyclamine, which is the predominant agent found in plasma and extravascular tissues (366,367). This macrolide demonstrates high and prolonged tissue concentrations, allowing once-daily dosing. The mechanism of action is identical to that of the other macrolides. The result is bacteriostatic activity against logarithmically growing microorganisms and may include bactericidal activity against bacteria in a static growth phase. Dirithromycin, like azithromycin, does not inhibit cytochrome P450 enzymes and thus does not cause clinically important drug–drug interactions, although its gastrointestinal side effects are similar to those of other macrolides (362,368). Macrolides appear to be able to suppress the initiation of mRNA synthesis (180) and thereby inhibit the production of biofilm (135,138), exoenzymes (131,132), and other such virulence factors by a diverse group of pathogens, including P. aeruginosa (131,269). If this proves to be clinically effective in chronic infections, a once-daily dosing that achieves high and prolonged tissue concentrations and has no significant drug–drug interactions will be extremely useful.
Telithromycin. Telithromycin is a ketolide (371) in which the C11/C12 carbamate residue includes a butyl chain linking an imidazole ring and a pyridine ring (358–360). The most important factors in terms of the structure and activity of telithromycin are the lack of the neutral sugar cladinose in position C3 as well as a C11/C12 carbamate group, which together markedly increase the affinity of telithromycin for its microbial target, the 23S ribosomal drug-binding pocket (352,353,369). Telithromycin interacts with the 23S rRNA portion of the 50S subunit in the upper portion of the peptide exit channel close to the peptidyl transferase center and prevents the peptide chain from passing through the peptide exit channel (352,353,369). The increased affinity of telithromycin for the 23S rRNA is seen even in macrolide-resistant strains (370) and also results in concentration-dependent bactericidal activity and a prolonged post–antimicrobial effect against important respiratory tract pathogens. The microbiologic spectrum of activity for telithromycin includes S. pneumoniae, S. pyogenes, H. influenzae, Moraxella catarrhalis, Legionella species, Mycoplasma pneumoniae, and Chlamydia pneumoniae, which suggest that telithromycin will play an important clinical role in the empirical treatment of community-acquired respiratory tract infections (371). The pharmacokinetic profile of telithromycin demonstrates that this drug can be administered once daily without regard for meals and requires no dose reduction in elderly patients or those having hepatic impairment (372). Telithromycin is well absorbed after oral administration, rapidly penetrates into respiratory tissues and fluids, and is highly concentrated within white blood cells. Integration of pharmacokinetic and pharmacodynamic properties reveals that telithromycin has a high AUC:MIC ratio compared with macrolide antimicrobial agents, which results in enhanced efficacy. Resistance, although rare to date, can occur (356). Finally, telithromycin is well tolerated and has a low propensity for drug interactions.
Clindamycin. Clindamycin is a member of the lincosamides, which are chemically unrelated to the macrolides (354). Lincosamides consist of an amino acid linked to an amino sugar. Clindamycin is a 7-deoxy-7-chloro derivative of lincomycin, the first member of the lincosamide class. These two agents, like the macrolides, act on the peptidyltransferase center of the 50S subunit of bacterial ribosomes (134). Clindamycin may exhibit in vitro bactericidal activity against susceptible microorganisms such as S. aureus and B. fragilis, but its in vivo activity is considered bacteriostatic. The reason for this species-specific bactericidal activity is unknown but may reflect differences in the apoptotic mechanisms of these microorganisms.
Streptogramins. The streptogramin group of antibiotics includes the mikamycins, the pristinamycins, the oestreomycins, and the virginiamycins. These compounds are classified into two main groups: polyunsaturated cyclic peptidolides and cyclic hexadepsipeptides. Both groups possess a wide variety of chemical functions. Quinupristin/dalfopristin (Synercid) is a semisynthetic antibiotic consisting of two water-soluble streptogramin components: pristinamycin IA, a peptidic macrolactone, and pristinamycin IIA, a polyunsaturated macrolactone (373–375). These two macrolactones are modified to be water-soluble and together demonstrate synergistic and concentration-independent lethal activity against gram-positive pathogens, including S. aureus, in contrast to the individual components, which are only inhibitory (376). This bactericidal effect is seen clinically as well, as evidenced by the successful treatment of bacterial endocarditis. It is thought that this activity is related to irreversible binding to ribosomes (377). The 70S ribosomal subunit appears to be the target, with binding, closing, or narrowing the extrusion channel. This combination also demonstrates a postantibiotic effect (378). Moreover, each component diffuses throughout cardiac vegetations and is more concentrated in these vegetations than in cardiac tissue. The peptide macrolactone is distributed homogeneously throughout each vegetation, while the polyunsaturated macrolactone reaches the core, with a gradient of decreasing concentrations from the periphery.
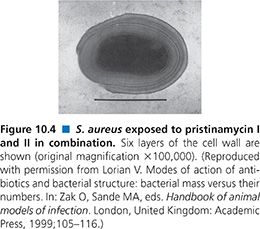
Exposure of S. aureus to quinupristin/dalfopristin results in two major cell alterations: an increase in cell size and an increase in the thickness of the cell wall. Some cells exhibit multilayered cell walls with as many as six layers (92) (Fig. 10.4), which is considerably thicker than the two- or three-layer cell wall that has been seen with exposure of S. aureus to chloramphenicol (94). This alteration is likely to be a stringent response.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


