, Arthur H. Cohen2, Robert B. Colvin3, J. Charles Jennette4 and Charles E. Alpers5
(1)
Department of Pathology, Microbiology and Immunology, Vanderbilt University Medical Center, Nashville, Tennessee, USA
(2)
Department of Pathology and Laboratory Medicine, Cedars-Sinai Medical Center, Los Angeles, California, USA
(3)
Department of Pathology Harvard Medical School, Massachusetts General Hospital, Boston, Massachusetts, USA
(4)
Department of Pathology and Laboratory Medicine, University of North Carolina, Chapel Hill, North Carolina, USA
(5)
Department of Pathology, University of Washington, Seattle, Washington, USA
Abstract
Minimal change disease (MCD) and focal segmental glomerulosclerosis (FSGS) are both common causes of the nephrotic syndrome. Minimal change disease accounts for greater than 90 % of cases of nephrotic syndrome in children vs. 10–15 % of adults with nephrotic syndrome [1]. Focal segmental glomerulosclerosis has been increasing in incidence in the United States in both African Americans and in Hispanics, in both adult and pediatric populations [2–4]. It is now the most common cause of nephrotic syndrome in adults in the USA. Patients with FSGS may have hypertension and hematuria. Serologic studies, including complement levels, are typically within normal limits in both MCD and FSGS.
Introduction/Clinical Setting
Minimal change disease (MCD) and focal segmental glomerulosclerosis (FSGS) are both common causes of the nephrotic syndrome. Minimal change disease accounts for greater than 90 % of cases of nephrotic syndrome in children vs. 10–15 % of adults with nephrotic syndrome [1]. Focal segmental glomerulosclerosis has been increasing in incidence in the United States in both African Americans and in Hispanics, in both adult and pediatric populations [2–4]. It is now the most common cause of nephrotic syndrome in adults in the USA. Patients with FSGS may have hypertension and hematuria. Serologic studies, including complement levels, are typically within normal limits in both MCD and FSGS.
Pathologic Features
Light Microscopy
Minimal change disease shows normal glomeruli by light microscopy (Fig. 4.1). In FSGS, sclerosis involves some, but not all, glomeruli (focal), and the sclerosis affects a portion of, but not the entire, glomerular tuft (segmental) (Fig. 4.2) [1, 5–7]. Sclerosis is defined as increased matrix with obliteration of the capillary lumen. Uninvolved glomeruli in FSGS show no apparent lesions by light microscopy; FSGS may also entail hyalinosis, caused by insudation of plasma proteins, producing a smooth, glassy (hyaline) appearance (Fig. 4.3). Adhesions (synechiae) of the capillary tuft to Bowman’s space are a very early sclerosing lesion.
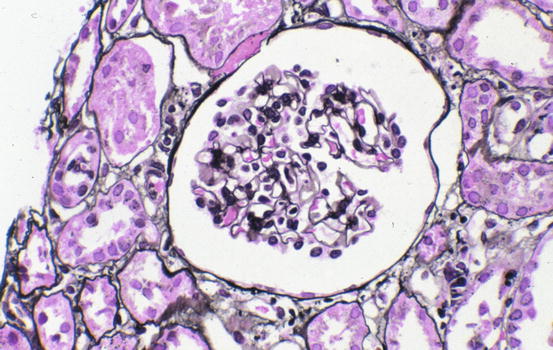
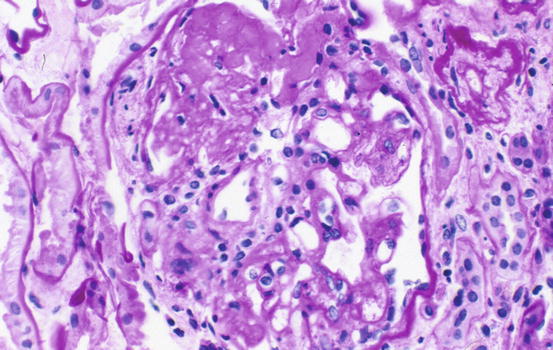
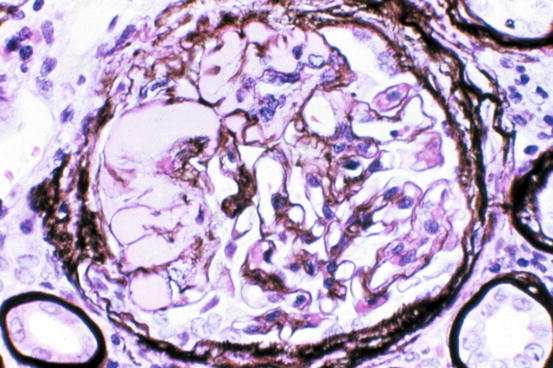
Fig. 4.1
Normal glomerulus in minimal change disease (Jones silver stain)
Fig. 4.2
Glomerulosclerosis in focal and segmental pattern in focal segmental glomerulosclerosis, not otherwise specified (FSGS NOS) type [periodic acid-Schiff (PAS)]
Fig. 4.3
Extensive hyalinosis and sclerosis in FSGS NOS type (Jones silver stain)
Focal segmental glomerulosclerosis is diagnosed when even a single glomerulus shows segmental sclerosis. Therefore, samples must be adequate to detect these focal and segmental lesions. A biopsy with only ten glomeruli has a 35 % probability of missing a focal (10 % involved) lesion, decreasing to 12 % if 20 glomeruli are sampled [8]. The juxtamedullary region also should be included in the biopsy for optimal sampling, because that is where FSGS starts [5]. Multiple-step sections should also be examined to detect the focal segmental lesions. As the disease process progresses, more glomeruli are involved more extensively, so that as the disease approaches end stage, all glomeruli may be involved with sclerosis, and the segmental sclerosis may then be extensive and global or near-global.
Global glomerulosclerosis, in contrast to the segmental lesion, is not of special diagnostic significance in diagnosing FSGS. Globally sclerotic glomeruli may be normally seen at any age. In children, less than 1 % global sclerosis is expected. The extent of global sclerosis increases with aging. Smith et al. [9] proffered the formula for normal percent of global sclerosis in adults of up to half the patient’s age, minus 10, for example, up to 30 % by age 80.
Morphologic variants of sclerosis appear to have differing prognosis (see below) [12–16]. The most common type of FSGS, FSGS not otherwise specified (NOS), has no specific distinguishing features and is characterized by segmental sclerosis, defined as increased matrix and obliteration of capillary lumina, often with hyalinosis in the absence of underlying immune complexes or other indicators of a secondary etiology by complete immunofluorescence/electron microscopy (IF/EM) evaluation; FSGS NOS is diagnosed by exclusion of specific subtypes as follows (Fig. 4.4): Collapsing variant of FSGS is characterized by collapse of the capillary loops and overlying podocyte proliferation and has a particularly ominous prognosis (Fig. 4.5) [17, 18]. Even just one glomerulus with collapsing lesion is sufficient, we propose, to classify the process as collapsing variant FSGS. In the absence of collapsing lesions, the location of lesions, either peripheral or hilar, has significance as follows: The glomerular tip lesion is defined as sclerosis only affecting the tubular pole of the glomerulus, with adhesion of the tuft to the proximal tubule outlet (Fig. 4.6). There is often associated endocapillary hypercellularity and intracapillary foam cells [12, 19, 20].
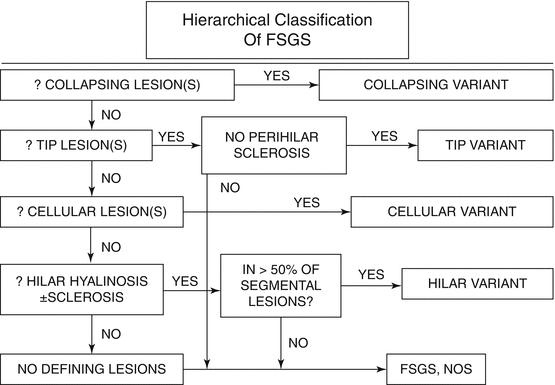
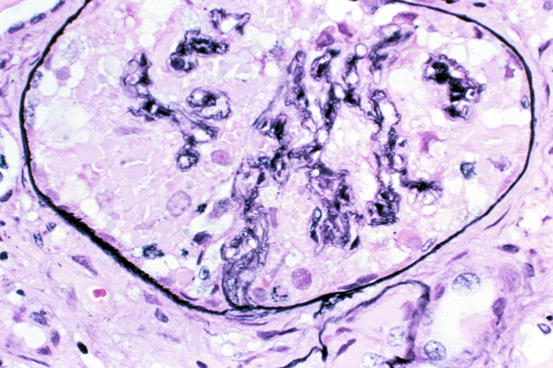
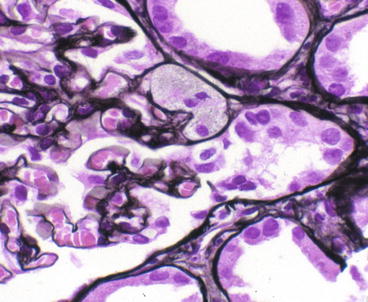
Fig. 4.4
Hierarchical classification schema for FSGS variants
Fig. 4.5
Collapse of glomerular tuft with overlying epithelial cell hyperplasia in collapsing-type FSGS (Jones silver stain)
Fig. 4.6
Adhesion and endocapillary foam cells at proximal tubular pole in tip variant of FSGS (Jones silver stain)
The cellular variant of FSGS is characterized by segmental proliferative podocyte reaction associated with early sclerosis and/or endocapillary hypercellularity and often intracapillary foam cells [14]. It does not, per definition, involve the tubular pole. Predominantly hilar lesions, that is, more than half of sclerotic glomeruli with sclerosis with associated hyaline at the hilar pole, in the absence of collapsing lesions, have been proposed to represent a response to reduced renal mass and may also be associated with secondary FSGS seen with arterionephrosclerosis [10, 11].
Vascular sclerosis may be prominent late in the course of FSGS and is proportional to glomerular sclerosis. Tubular atrophy is often accompanied by interstitial fibrosis, proportional to the degree of scarring in the glomerulus. In HIV nephropathy and collapsing glomerulopathy, tubular lesions are disproportionally severe, with cystic dilation and a more prominent infiltrate [21].
The presence of acute interstitial nephritis (i.e., edema, interstitial infiltrate of lymphocytes, plasma cells, and often eosinophils) and apparent MCD glomerular lesion (i.e., complete foot process effacement, no light microscopic lesions) suggests a drug-induced hypersensitivity etiology, in particular nonsteroidal anti-inflammatory drug (NSAID)-related injury.
Surrogate markers of unsampled FSGS have been sought to suspect FSGS even when sclerosed glomeruli are not detected. Abnormal glomerular enlargement (see below) appears to be an early indicator of the sclerotic process preceding overt sclerosis [22]. Tubulointerstitial fibrosis in a young patient without evident glomerular sclerosis in the biopsy sample could also indicate possible unsampled FSGS.
Immunofluorescence Microscopy
There are no immune complex deposits in either MCD or FSGS. In FSGS, there may be nonspecific entrapment of immunoglobulin M (IgM) and C3 in sclerotic areas or areas where mesangial matrix is increased. IgM staining without deposits by EM does not appear to have specific diagnostic, prognostic, or etiologic significance [23].
Electron Microscopy
Electron microscopy shows foot process effacement, vacuolization, and microvillous transformation of epithelial cells in both MCD and FSGS. In MCD, foot process effacement is typically extensive (Fig. 4.7). Foot process effacement is often not complete in FSGS [23]. However, the extent of foot process effacement does not allow precise distinction between the two disease processes. In secondary FSGS, foot process effacement is generally less than in idiopathic forms. Thus, less than about 50 % foot process effacement is suggestive of a secondary etiology of the segmental sclerosis rather than idiopathic FSGS [24]. In HIV-associated nephropathy (HIVAN), there are reticular aggregates in endothelial cell cytoplasm.
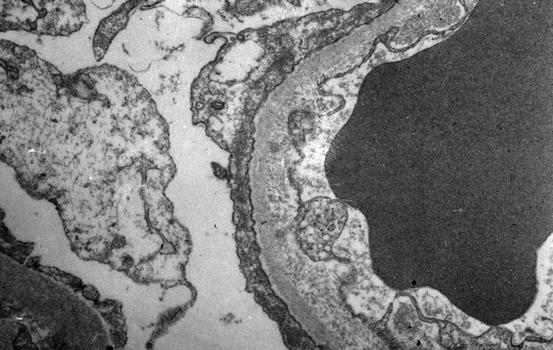
Fig. 4.7
Extensive foot process effacement is present in minimal change disease and also in FSGS. No immune complexes are present (electron microscopy)
Etiology/Pathogenesis
The pathogenesis of MCD appears related to abnormal cytokines, which only affect glomerular permeability and do not promote sclerogenic mechanisms. Urinary CD80 is increased in MCD but not in FSGS patients, suggesting that CD80 could be a marker or possibly contribute to pathogenesis of MCD [25]. CD80 is a transmembrane protein that provides a co-stimulatory signal for T cell activation and may be induced on podocytes in response to activated T cells or other injury. Minimal change disease has been associated with drug-induced hypersensitivity reactions, bee stings, Hodgkin’s disease, and other venom exposure, implicating immune dysfunction as an initiating factor.
Glomerular hypertrophy may be a marker of early FSGS. Glomerular enlargement precedes overt glomerulosclerosis in FSGS [22]. Patients with abnormal glomerular growth on initial biopsies that did not show sclerotic lesions subsequently developed overt glomerulosclerosis, documented in later biopsies. A cutoff of glomerular area larger than 50 % more than normal for age indicated increased risk for progression. Of note, glomeruli grow until approximately age 18 years; so age-matched controls must be used in the pediatric population. Since tissue processing methods may influence the size of structures in tissue, it is imperative that each laboratory determines normal ranges for this parameter.
Recurrence of FSGS in the transplant has also shed light on its pathogenesis [26]. Most recurrences occur within the first months after transplantation, but recurrence may be immediate. Foot process effacement is present when proteinuria recurs and precedes the development of sclerosis, typically by weeks to months. The pattern of FSGS generally, but not always, is that of the original disease [27]. A circulating factor has been identified in patients with recurrent FSGS, which induces increased ex vivo glomerular permeability, and also a mild increase in proteinuria when injected in rats [28]. Plasmapheresis induced remission in some patients with recurrent FSGS, but more often relapse occurred when plasmapheresis was stopped. During the stage of foot process effacement in recurrent FSGS, but not in native kidney MCD, activated CD44-positive parietal epithelial cells were detected, both in a parietal and in a visceral location, suggesting that parietal epithelial cells are activated and perhaps migrating to sites of injury [29]. Parietal epithelial cells may act as stem cells to replenish podocytes, migrating along Bowman’s capsule or adhesions to the glomerular capillary tuft [30]. Whether these cells with parietal epithelial cell markers and visceral location are reparative or profibrotic is not determined [31, 32].
Differentiation markers of podocytes, including the Wilms’ tumor WT-1 protein, podocalyxin, and synaptopodin, are retained when proteinuria is caused by MCD or membranous glomerulopathy but disappeared (or were decreased in the case of synaptopodin) in the collapsing variant of FSGS or HIV-associated nephropathy, with lesser changes in typical FSGS, with gain of markers of activated parietal epithelial cells [29, 33, 34]. These observations point to a dysregulated phenotype of the visceral epithelial cell in the pathogenesis of the collapsing and HIV-associated forms of FSGS, perhaps with migration of activated parietal epithelial cells, or with transdifferentiation of the podocytes to a parietal phenotype [29, 33, 34].
Studies of the molecular biology of the podocyte and identification of genes mutated in rare familial forms of nephrotic syndrome or FSGS, e.g., ACTN4, NPHS2, TRPC–6, PLCE1, INF–2, WT1, CD2AP, LAMB2, and NPHS1, have given important new insights into mechanisms of progressive glomerulosclerosis [35, 36]. Congenital nephrotic syndrome of Finnish type (CNSF) is caused by mutations in nephrin (NPHS1) [37]. The common mutations in this syndrome are called fin major and fin minor. Nephrin localizes to the slit diaphragm over the podocyte and is tightly associated with CD2-associated protein (CD2AP) [38]. Nephrin functions as a zona occludens-type junction protein and along with CD2AP plays a crucial role in receptor patterning and cytoskeletal polarity. Nephrin also has signal transduction functions. Some patients with CNSF develop nephrotic syndrome after transplantation, if the original disease was due to fin major mutations, which leads to complete loss of nephrin, with antibody to nephrin detected in the patient. Mice engineered to be deficient in CD2AP develop congenital nephrotic syndrome. Mutations of CD2AP, predicted to cause loss of function, have been detected in two adult patients with proteinuria and FSGS without a family history of the disease [39].
Autosomal dominant FSGS is caused by mutation in α-actinin-4, a key cytoskeletal protein (ACTN4) [40]. Patients with the ACTN4 mutation progress to end stage by age 30 years. Podocin, another podocyte-specific structural gene (NPHS2), is mutated in autosomal recessive FSGS with an early onset in childhood with rapid progression to end stage [41]. Importantly, patients with such podocin mutations are generally resistant to steroid therapy, whereas some patients with PLCE1 mutations may respond to steroids [36, 42]. Acquired FSGS also may involve alteration in expression of some of these key podocyte genes that confer increased susceptibility to injury and increased risk of FSGS. An allele variant of apolipoprotein L1 that is protective against trypanosomal disease has been linked to increased FSGS in African Americans [43]. The mechanisms for renal disease susceptibility remain unknown.
Additional observations underscore the importance of the interactions between the podocyte and the underlying basement membrane. Dystroglycan is an integral component of the GBM. Dystroglycan staining was decreased in patients with MCD and also in the proliferating epithelial cells in collapsing glomerulopathy [44, 45]. Dystroglycan expression was maintained in the nonsclerotic segments in FSGS NOS variant, suggesting that MCD and FSGS are indeed different disease processes and not different stages of one disease.
Germline mutations of the WT-1 (Wilms’ tumor) suppressor gene are found in Denys-Drash syndrome (a rare childhood disease with diffuse mesangial sclerosis, male pseudohermaphroditism, and a high risk of Wilms’ tumor, with mutations usually of exon 9) and in Frasier syndrome (a disease with FSGS, XY hermaphroditism, and a high risk of gonadoblastoma, with mutations of intron 9) [46]. Abnormal, lamellated basement membranes were observed in three patients with FSGS associated with Frasier syndrome, in whom studies for coexistent Alport syndrome were negative [47]. Abnormal splice variants of WT-1 have rarely been associated with nonsyndromal cases of FSGS [48].
Clinicopathologic Correlations
Minimal change disease patients typically respond to corticosteroids, with excellent long-term prognosis. In contrast, FSGS usually results in progressive decline of GFR. The finding of mesangial hypercellularity was proposed to indicate a subtype of primary MCD with poorer prognosis and increased risk for development of FSGS. However, several series have failed to confirm a definite clinical correlation of this morphologic variant. Thus, diffuse mesangial hypercellularity does not appear to impart a specific prognostic significance and is rather regarded as a manifestation of an earlier stage of disease.
The proposed morphologic variants of FSGS appear to have prognostic significance (Table 4.1) [10, 11, 16]. Collapsing FSGS has a poor prognosis with rapid loss of renal function and virtually no responsiveness to corticosteroids alone [18]. Series of patients with collapsing FSGS show a strong preponderance of African Americans, and most patients were adults. FSGS in African Americans is tightly linked to risk allele variants of the apolipoprotein A1 gene [43]. Evidence of parvovirus infection was increased in patients with collapsing glomerulopathy compared to controls, usual-type FSGS and HIVAN [49]. The drug pamidronate and other bisphosphonates also have been linked to the development of collapsing glomerulopathy [41]. Collapsing lesions also have been seen in the transplant, linked to cyclosporine toxicity, with other causes of severe ischemia in lupus patients, and with exogenous interferon therapy [50].
Table 4.1




Working classification of focal segmental glomerulosclerosis (FSGS)
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
