Mesenchymal Neoplasms
BENIGN MESENCHYMAL TUMORS
Fibromatosis is an infiltrating, histologically low grade spindle cell neoplasm composed of fibroblastic cells and variable amounts of collagen.
Patients with mammary fibromatosis range from ages 13 to 80 at diagnosis, averaging 37 to 49 years in three reported series (1,2,3). They usually present with a palpable, firm, painless mass that may suggest carcinoma on clinical examination. Bilateral fibromatosis is very uncommon. Mammography reveals a stellate tumor that may be indistinguishable from carcinoma (1,3,4,5,6). Calcifications are rarely formed in mammary fibromatosis, but they may be present in a coexisting benign proliferative lesion, such as sclerosing adenosis that has been engulfed by the tumor. Rarely, the tumor may be nonpalpable and initially detected by mammography or ultrasonography (7). Antecedent injury or surgery has been reported at the site of fibromatosis in some patients, and an association with breast augmentation implants has been reported in several cases (2,8). Tumor size averages 2.5 to 3.0 cm (2,3).
A single lesion may have varied growth patterns, a feature that complicates the diagnosis of fibromatosis in a needle core biopsy specimen. The microscopic components of the tumor are spindle cells and collagen. Areas in which the collagenous element is accentuated have a keloidal appearance (Fig. 23.1). More commonly, the lesion is composed of a moderately cellular spindle cell proliferation in which there is modest collagen deposition. Mitotic figures are very infrequent or undetectable. In most tumors, the cells have a small, pale, oval or spindly nuclei with little pleomorphism (Fig. 23.2). The tumor cells are usually distributed in broad sheets, sometimes in a storiform configuration or in the form of interlacing bundles. Actin-or CD 34-positive myofibroblasts are usually inconspicuous. In addition to varied amounts of collagenization, the stroma sometimes has localized myxoid areas. Focal lymphocytic infiltrates are found in nearly half of the tumors, especially at the periphery (Fig. 23.3). Regardless of how well demarcated the lesions seem to be grossly, all have at least some stellate, invasive extensions into the surrounding fat and glandular parenchyma. It is usually possible to find ducts and lobules engulfed by these extensions at the periphery of the tumor (Fig. 23.4). The differential diagnosis of mammary fibromatosis includes scarring from trauma or prior surgery, metaplastic spindle-cell carcinoma, and other spindle cell mesenchymal neoplasms, including phyllodes tumor.
Rare examples of mammary fibromatosis have been associated with familial adenomatous polyposis (FAP), a condition in which somatic fibromatosis (desmoid tumor) frequently occurs (9). FAP is the result of germ-line mutations in the adenomatous polyposis coli or APC gene located on chromosome 5q. The APC gene product, the APC tumor suppressor protein, regulates β-catenin, a component of the cadherin cell-to-cell adhesion system and of the Wnt pathway (10). Altered regulation of β-catenin results in nuclear accumulation of β-catenin protein. In a study of 32 examples of sporadic mammary fibromatosis and 1 FAP-associated tumor, Abraham et al. (11) found β-catenin accumulation in tumor cell nuclei in 82% of the tumors but not in the nuclei of normal stromal cells or epithelial cells. In addition to this immunohistochemical evidence of nuclear translocation of β-catenin, most of the lesions had genetic alterations in the APC/β-catenin pathway, consisting of β-catenin gene mutation (15 cases) and mutation of 5q (11 cases). If confirmed and found to be specific for fibromatosis of the breast, whether sporadic or FAP-associated, these results suggest that the β-catenin immunostain could be useful for distiguishing fibromatosis from other spindle cell lesions of the breast.
Recommended treatment is wide local excision. The frequency of local recurrence ranges from 21% to 27% (1,2,3). Histologic features, such as cellularity, mitotic activity, and cellular pleomorphism are not helpful for predicting recurrence. Although the risk of recurrence is higher in patients
with documented positive margins, recurrences have been observed in cases with apparently negative margins (3).
with documented positive margins, recurrences have been observed in cases with apparently negative margins (3).
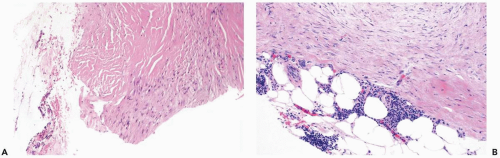 Figure 23.1 Fibromatosis, keloidal. A. This needle core biopsy specimen shows an area of dense collagenous tissue adjacent to a more cellular component of the lesion. The slit-shaped spaces in the keloidal tissue create a resemblance to pseudoangiomatous stromal hyperplasia. B. Fibromatosis with peripheral lymphoid aggregates. |
Fibrous tumor (focal fibrous disease) is a discrete breast tumor composed of collagenized mammary stroma (12). Fibrous tumor is a disease of premenopausal women. Mammography reveals an area of density with borders varying from irregular to smooth. Calcifications are not a feature of fibrous tumor. This benign stromal proliferation is treated by local excision.
Harvey et al. (13) studied 14 patients with “fibrous nodules” that appear to correspond histologically to fibrous tumors. Ten patients (71%) were premenopausal. Three of the four postmenopausal women were receiving hormone replacement therapy. The lesions measured 0.6 to 3.5 cm with a mean size of 1.8 cm. Twelve women had nonpalpable mammographically detected tumors. One woman had synchronous bilateral nonpalpable lesions. In mammograms, 11 of 13 nonpalpable tumors were round or oval, 6 of the 11 had circumscribed margins, and 5 had indistinct borders. Two tumors had irregular shapes and spiculated margins.
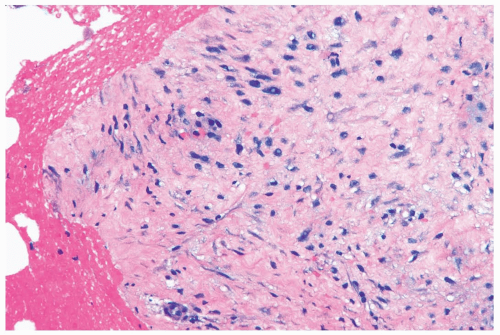 Figure 23.2 Fibromatosis. A needle core biopsy specimen showing average cellularity and slight stromal edema. The tumor cells have uniform round-to-oval nuclei. |
Histologic examination reveals normal appearing, collagenous stroma that contains markedly decreased or absent ductal and lobular elements (12,14). The findings in a needle core biopsy specimen are nonspecific and are usually reported as “fibrosis” (Fig. 23.5). Capillaries, other vascular structures, and nerves are very sparse; perivascular and perilobular inflammatory infiltrates are absent. Cysts, apocrine metaplasia, sclerosing adenosis, pseudoangiomatous stromal hyperplasia, and duct hyperplasia are not features of fibrous tumor.
Pseudoangiomatous stromal hyperplasia (PASH) can be mistaken for angiosarcoma. The term pseudoangiomatous was proposed to emphasize that the histologic pattern mimics but does not constitute a vasoformative proliferation. PASH is a tumor formed by myofibroblasts with variable expression of myoid and fibroblastic features. Glandular hyperplasia is sometimes also present. Some
examples of tumor-forming PASH have been misclassified as mammary hamartomas (15).
examples of tumor-forming PASH have been misclassified as mammary hamartomas (15).
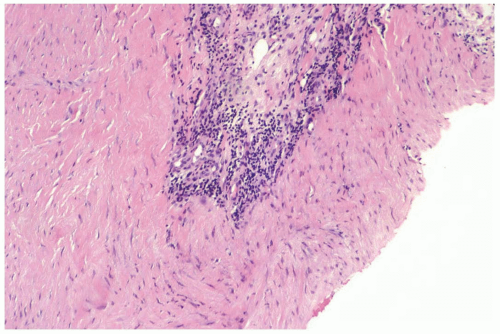 Figure 23.3 Fibromatosis. A perivascular lymphocytic infiltrate is apparent in this needle core biopsy specimen. |
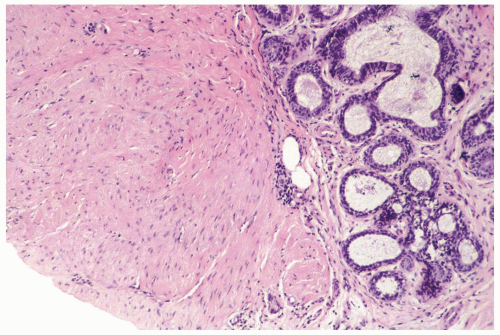 Figure 23.4 Fibromatosis. The tumor invades up to a lobule in this part of a needle core biopsy specimen. |
With rare exceptions, reported examples of tumor-forming PASH have been in women. PASH is a frequent incidental component of gynecomastia, having been found in 44 of 93 consecutive male breast biopsies (47.4%); 43 of the 44 male specimens with PASH were from patients with gynecomastia (16). The age at diagnosis in women ranges from the teens to the mid-50s, with a median age in the mid- to late 30s (17,18,19). Almost all patients have been premenopausal. Most women have a palpable, painless, unilateral mass that is firm or rubbery. Any part of the breast can be affected, including the nipple-areola complex (20). Palpable tumors average 5 cm in diameter. Skin necrosis has been observed in patients who have massive breast enlargement due to PASH during pregnancy.
PASH has been detected by mammography in patients who were asymptomatic (21,22). The lesion presents as a mass without calcification. The borders are usually smooth, but a minority of the tumors are spiculated or have ill-defined margins, sometimes obscured by surrounding tissue. Ultrasound reveals a well-defined, usually hypoechoic mass (22,23). Clinically asymptomatic PASH detected by mammography may occur in postmenopausal patients, whereas palpable lesions are almost always found in premenopausal women or in postmenopausal women who have been treated with hormone replacement therapy.
The tumors are composed of intermixed stromal and epithelial elements in which lobular and duct structures are separated by an increased amount of stroma. Collagenization of intralobular stroma and duct attenuation producing fibroadenoma-like features are common. Nonspecific proliferative epithelial changes include mild hyperplasia of duct and lobular epithelium, often with some accentuation of myoepithelial cells, and apocrine metaplasia with or without cyst formation.
The most striking histologic finding is a complex pattern of anastomosing slit-shaped spaces in the intralobular as well as interlobular stroma (Fig. 23.6). Myofibroblasts are present singly and intermittently at the margins of the spaces that resemble endothelial cells. The nuclei of most of the myofibroblasts are attenuated, lack atypia, and do not show mitotic activity. Rarely, some of these cells are enlarged and have noticeably hyperchromatic nuclei. Also present in the stroma are round or oval blood-containing true blood vessels lined by endothelial cells (Figs. 23.6, 23.7).
The myofibroblasts may accumulate in distinct bundles or fascicles in PASH (Figs. 23.8, 23.9). The most pronounced examples of this cellular form of PASH have a growth pattern reminiscent of a myofibroblastoma. This is especially the case when the myofibroblasts have abundant cytoplasm and PASH occurs as a localized tumor rather than as a diffuse process. Myofibroblastoma and PASH are related conditions, representing the extremes of a spectrum of lesions, sharing a common histogenesis in the myofibroblasts. An atypical form of PASH has pleomorphic nuclei and infrequent mitotic figures (Fig. 23.10).
Basement membrane material is not present around the slit-like spaces in PASH. Myofibroblasts defining the pseudoangiomatous spaces typically exhibit strong immunoreactivity for vimentin and CD34. They are variably reactive for actin, calponin and desmin and show no immunoreactivity for vascular markers. The nuclei of myofibroblasts in PASH are sometimes immunoreactive for progesterone receptor (17,24). Estrogen receptor is absent or only weakly present when the tissues are examined by immunohistochemistry (17,24).
Pseudoangiomatous stromal hyperplasia, which forms a clinically palpable tumorous mass, appears to be an exaggerated manifestation of physiologic changes commonly encountered microscopically. Ibrahim et al. (18) found microscopic foci of PASH in 23% of 200 consecutive breast specimens obtained for benign or malignant conditions. Eighty-nine percent of the patients were younger than age 50. The majority of these specimens also exhibited epithelial hyperplasia, sometimes including secretory changes in lobules.
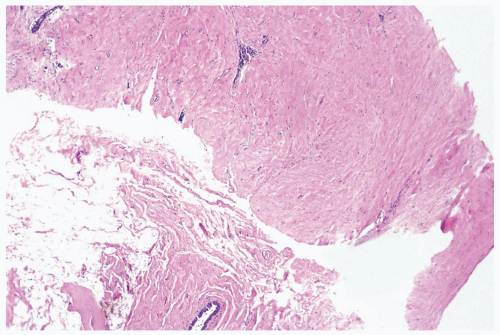 Figure 23.5 Fibrous tumor. The upper sample in this needle core biopsy specimen shows a broad expanse of collagenous tissue from the tumor. In the lower sample, a duct is surrounded by normal fibrofatty tissue. |
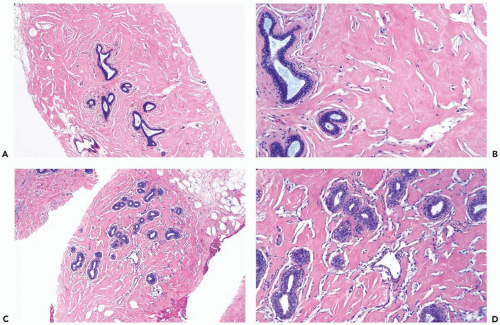 Figure 23.6 Pseudoangiomatous stromal hyperplasia. Needle core biopsy specimens from two patients. A, B. In this instance, myofibroblasts are inconspicuous, and the slit-shaped spaces are largely unconnected. Collagen fibrils extend across some of the spaces. C, D. This very pronounced pseudoangiomatous proliferation involves a lobule and the surrounding stroma. |
Most patients have remained well after excisional biopsy, but ipsilateral recurrences have occurred in some cases. Bilateral involvement, an infrequent occurrence, may be simultaneous or metachronous. Anti-estrogen therapy may be beneficial in some cases (25,26).
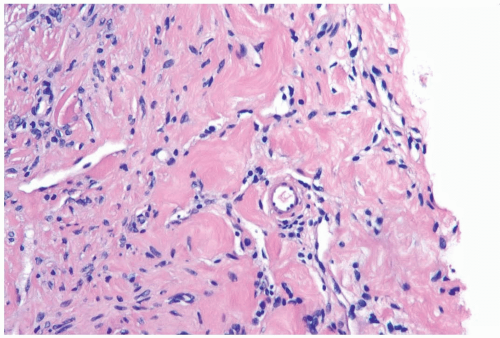 Figure 23.7 Pseudoangiomatous stromal hyperplasia. Small blood vessels are distributed among the pseudoangiomatous spaces in this needle core biopsy specimen. |
Myofibroblastoma is a benign tumorous proliferation of myofibroblasts. These spindle-shaped or fusiform mesenchymal cells have cytoplasmic actin-like microfilaments measuring 5 to 7 nm in diameter with focal dense bodies and pinocytotic vesicles when studied by electron microscopy. Desmosomes are absent or poorly formed between myofibroblasts. Myofibroblasts exhibit variable expression of actin and CD34 in various proliferative and neoplastic conditions. According to Chauhan et al. (27), in the normal breast and in the presence of most proliferative lesions, myofibroblasts are CD34 positive and β-actin negative, whereas in invasive carcinoma they are CD34 negative and alpha-actin positive. In the presence of intraductal carcinoma, expression of CD34 and alpha-actin in the surrounding breast is related to the grade of the carcinoma, with loss of CD34 expression greatest in high grade lesions.
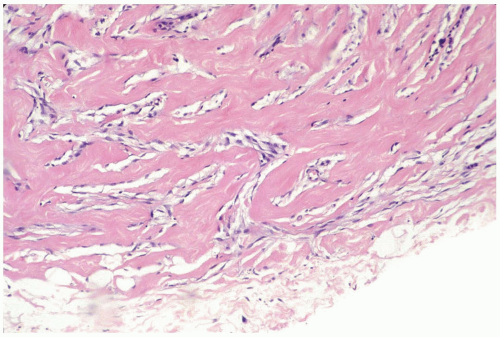 Figure 23.8 Pseudoangiomatous stromal hyperplasia. The lesion shown in this needle core biopsy specimen has a circumscribed border. The presence of myofibroblastic nuclei in some of the spaces is an early phase in the development of the fascicular pattern. |
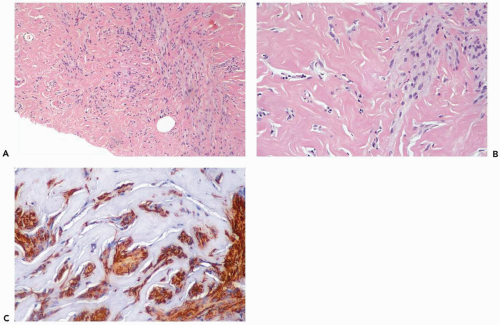 Figure 23.9 Pseudoangiomatous stromal hyperplasia, fascicular. A. Myofibroblasts have formed distinct bundles that are distributed in the pseudoangiomatous pattern in this needle core biopsy specimen. B. Another portion of the specimen showing pseudoangiomatous and fascicular elements. C. The myofibroblasts are immunoreactive for actin (immunoperoxidase). |
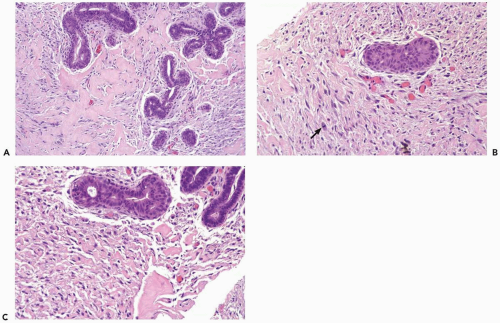 Figure 23.10 Atypical pseudoangiomatous stromal hyperplasia, fascicular. A-C. This needle core biopsy specimen was obtained from a 12-cm tumor in a 29-year-old woman. The stroma has occasional mitotic figures (arrow) and is focally very cellular. The pseudoangiomatous pattern is maintained in cellular foci. Ductal hyperplasia is also evident. |
The patient typically presents with a solitary unilateral mass in the breast. The median age is about 65 years. Many patients reported to have myofibroblastoma have been men, but the lesion also occurs in women (28). Radiographically, the tumors are homogeneous, lobulated, well circumscribed, and lack microcalcifications (29,30). The average diameter of the tumor is about 2 cm with most smaller than 4 cm. Size extremes include one lesion that measured 0.9 cm (28) and one 10 cm (31). Excisional biopsy is adequate treatment in most cases, and local recurrence is infrequent.
The classic type of myofibroblastoma is composed of bundles of slender, bipolar, uniform, spindle-shaped cells, typically arranged in clusters that are separated by broad bands of hyalinized collagen (32) (Fig. 23.11). Multinucleated giant cells are uncommon and mitotic figures are sparse or undetectable. Fat cells, ducts, and lobules are present in a minority of lesions. Usually, these components appear to have been incorporated from the surrounding tissue, especially if the tumor displays invasive growth. Rarely, fat cells are dispersed separately or in small groups throughout the tumor or there is abundant fat suggestive of a lipomatous element. The term lipomatous myofibroblastoma has been suggested for the latter group of lesions (33). The distinction between lipomatous myofibroblastoma and spindle cell lipoma is not clear, however. Gene rearrangements have been reported to affect 13q and 16q in spindle cell lipomas (34), and they have also been detected in myofibroblastomas (35).
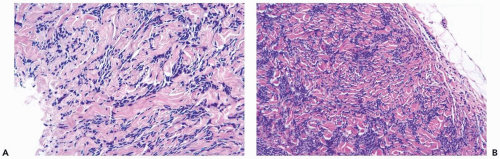 Figure 23.11 Myofibroblastoma. A. This needle core biopsy specimen shows the characteristic fascicles of myofibroblasts and intervening bands of collagen. B. The excised tumor had a circumscribed border. |
Some lesions have foci of leiomyomatous differentiation. A perivascular lymphoplasmacytic infiltrate is sometimes identified. The border of the tumor is usually circumscribed microscopically, but in a minority of cases the tumor has an invasive margin. The majority of myofibroblastomas are immunoreactive for vimentin, actin, and CD34. Many are also desmin positive. The tumors are not immunoreactive for cytokeratin or factor VIII and, only rarely, weakly reactive for S-100 protein.
Variant forms of myofibroblastoma have received little attention. In a collagenized myofibroblastoma, the spindle cells are distributed in diffusely collagenized stroma (Fig. 23.12). Irregular slit-like spaces formed between tumor cells and the stroma are reminiscent of PASH. The epithelioid variant features polygonal or epithelioid cells arranged in alveolar groups (Fig. 23.13), often mixed with classical myofibroblastoma elements. A cellular, infiltrating type of myofibroblastoma is formed by a dense proliferation of spindle-shaped neoplastic myofibroblasts with cytologic atypia. Collagenous bands may be absent in some parts of the lesion. These tumors tend to have infiltrative borders microscopically (Fig. 23.14). Myxoid myofibroblastoma also displays infiltrative growth (Fig. 23.15).
Granular cell neoplasms are derived from the Schwann cells of peripheral nerves. They occur throughout the body, with about 5% of them originating in the breast (36). The patient usually presents with a solitary firm or hard, painless mass, most often located in the upper and medial breast quadrants. On mammography, granular-cell tumor of the breast (GCTB) is difficult to distinguish from carcinoma (37,38), and coexistence of the two lesions has been described (39). GCTB typically forms a stellate mass lacking calcifications, but circumscribed lesions have been reported (37,38,40,41). Ultrasound usually reveals a solid mass with posterior shadowing suggestive of carcinoma (40,41). Benign GCTB is treated by wide excision. Local
recurrence may occur after incomplete excision. Less than 1% of all granular cell tumors, including mammary lesions, is malignant.
recurrence may occur after incomplete excision. Less than 1% of all granular cell tumors, including mammary lesions, is malignant.
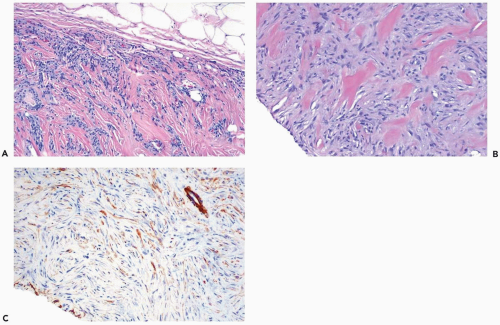 Figure 23.12 Myofibroblastoma, collagenized. A. Bundles of myofibroblasts are distributed between prominent bands of collagen. The tumor has a circumscribed border. B. Another tumor with dense collagen bands and myofibroblasts. C. CD34 reactivity in the tumor in (B) (immunoperoxidase). |
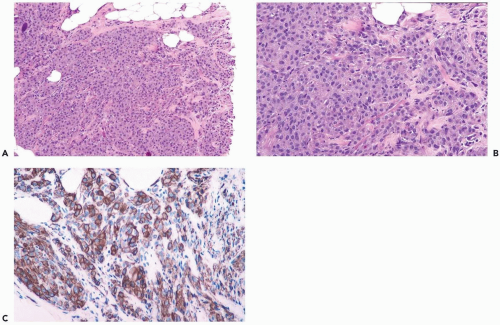 Figure 23.13 Myofibroblastoma, epithelioid. A, B. Epithelioid myofibroblasts form alveolar clusters and small bundles in this very cellular needle core biopsy specimen. This appearance could be mistaken for invasive carcinoma. C. The cells are strongly immunoreactive for smooth muscle actin (immunoperoxidase). |
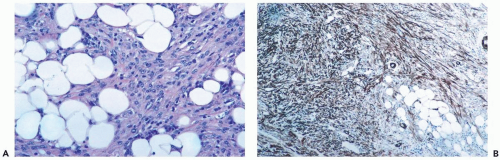 Figure 23.14 Myofibroblastoma, infiltrating. A. Tumor cells in fat might be mistaken for infiltrating carcinoma in this needle core biopsy specimen. B. The tumor cells are strongly immunoreactive for smooth muscle actin (immunoperoxidase). |
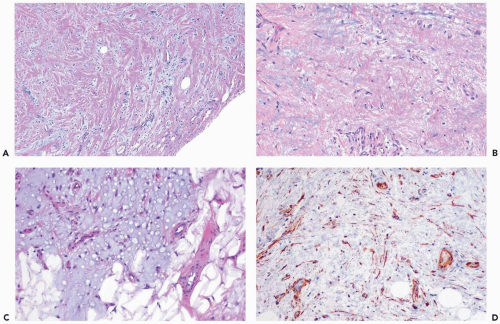 Figure 23.15 Myofibroblastoma, myxoid. A. A collagenized portion of the tumor. B. Basophilic myxoid material is evident in the tumor. C. Fully developed myxoid myofibroblastoma invading fat. D. The invasive myxoid portion of the tumor is immunoreactive for CD34 (immunoperoxidase). |
Granular-cell tumor of the breast is composed of compact nests or sheets of cells that contain eosinophilic cytoplasmic granules (Fig. 23.16). In some lesions there is a tendency to cytoplasmic vacuolization and clearing. The cytoplasmic granules are positive for periodic-acid-Schiff (PAS) and diastase resistant. The cells vary from polygonal to spindle in shape. Variable amounts of collagenous stroma are present. Nuclei are round to slightly oval with an open chromatin pattern. A modest amount of nuclear pleomorphism, occasional multinucleated cells, and rare mitoses may be found, but these features should not be interpreted as evidence of a malignant neoplasm. Small nerve bundles are sometimes seen in the tumor or in close association with the peripheral invasive border. Ducts and lobules are typically surrounded by the invasive tumor cells and incorporated into the lesion.
The differential diagnosis of GCTB includes apocrine carcinoma and histiocytic or granulomatous lesions. The presence of intraductal carcinoma, often of the comedo type, as well as marked cytologic pleomorphism, usually serves to identify apocrine carcinoma, but these features may be absent from a needle core biopsy specimen. The lesions can be distinguished with confidence by histochemical studies. GCTB is not reactive for epithelial markers and does not contain mucin. Strong, diffuse immunoreactivity for S-100 protein and CEA, which characterizes granular cell tumors, does not by itself distinguish these lesions from mammary carcinoma (38,42,43,44). A high proportion of granular cell tumors are reactive for vimentin, and they are estrogen and progesterone receptor negative. GCTB is immunohistochemically negative for histiocyte-associated antigens, such as β1-antitrypsin and muramidase (38,42).
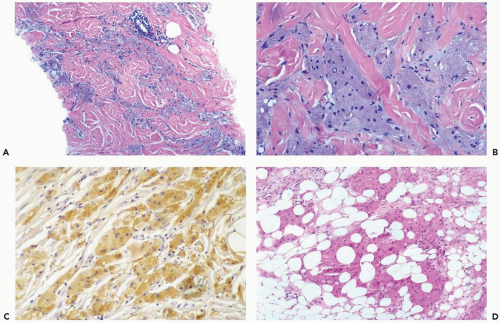 Figure 23.16 Granular-cell tumor. A, B. Bundles of elongated and polygonal tumor cells are present in collagenous breast stroma in this needle core biopsy specimen. This appearance could be mistaken for infiltrating apocrine carcinoma. C. The tumor cells are immunoreactive for S-100 protein (immunoperoxidase). D. Another lesion, in which a granular-cell tumor infiltrates fat and resembles apocrine carcinoma. |
Tumors of nerve and nerve sheath origin of the breast have usually been diagnosed as “neurilemomas” (45,46,47) or as schwannomas. The age range at diagnosis is 15 to 78 years, with most patients in their 30s and 40s. The lesion presents as a painless, well-defined mass. One schwannoma was an asymptomatic 7-mm lesion detected by mammography (47). Microscopic examination reveals the typical histologic features of a benign nerve sheath tumor, consisting of spindle cells in bundles, sometimes with nuclei
arranged in a palisading pattern (Antoni type A) (Fig. 23.17). Less cellular areas with thick walled blood vessels, the Antoni B pattern, may be present as well. Vascular thrombi, hyalinized blood vessels, cells with atypical nuclei, and xanthomatous areas are found in sclerotic schwannomas. The diagnosis of a benign peripheral nerve sheath tumor is supported by a positive immunohistochemical stain for S-100 protein, negative immunostains for CEA and actin, and the absence of mitotic activity. Complete excision provides adequate therapy.
arranged in a palisading pattern (Antoni type A) (Fig. 23.17). Less cellular areas with thick walled blood vessels, the Antoni B pattern, may be present as well. Vascular thrombi, hyalinized blood vessels, cells with atypical nuclei, and xanthomatous areas are found in sclerotic schwannomas. The diagnosis of a benign peripheral nerve sheath tumor is supported by a positive immunohistochemical stain for S-100 protein, negative immunostains for CEA and actin, and the absence of mitotic activity. Complete excision provides adequate therapy.
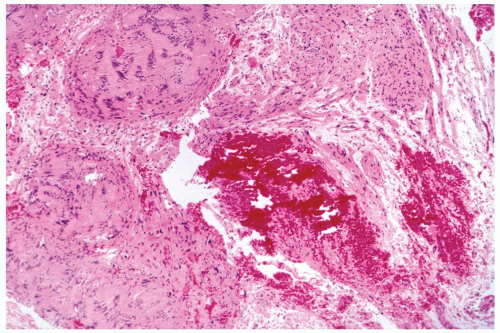 Figure 23.17 Schwannoma. The lesion has the Antoni type A pattern. Hemorrhage is at the site of a prior needle core biopsy procedure. |
Hamartoma of the breast occurs most often in premenopausal women, but it has been described in teenagers and in women in their 60s. The tumors have been as large as 17 cm, often resulting in substantial asymmetry. Mammography reveals a well-circumscribed, dense, round, or oval mass surrounded by a narrow lucent zone (48,49,50). Hamartomas of the breast were diagnosed in 16 of 10,000 mammography examinations reviewed by Hessler et al. (50). The ultrasonographic appearance of a mammary hamartoma is reported to be variable and not specific (51




Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
