Fig. 9.1
Histological subtypes of medulloblastoma. (a) Classic histology; (b) desmoplastic histology; (c) medulloblastoma with extensive nodularity (MBEN); (d) large cell/anaplastic (LC/A) histology.
Understanding the molecular biology underlying medulloblastoma is currently an area of intense interest among the pediatric neuro-oncology community. It is anticipated that knowledge gained from genomic and biological studies will translate to more accurate and consistent diagnoses, improved risk-stratification schemes, and the development and implementation of molecularly targeted therapies that are more effective and less toxic.
Molecular Genetics of Medulloblastoma: A Historical Perspective
Recurrent cytogenetic aberrations have been observed in medulloblastoma specimens for several decades [8]. The presence of an isochromosome 17q (i[17]q), essentially resulting in the net loss of one copy of the chromosome 17 p-arm and a net gain of one copy of the q-arm, is a signature event in medulloblastoma, found in up to 50 % of patient samples (Fig. 9.2a). Other chromosomal abnormalities that are commonly encountered include gains of chromosomes 1q and 7 and losses of chromosomes 6, 8p, 9q, 10q, 11, 16q, and X.
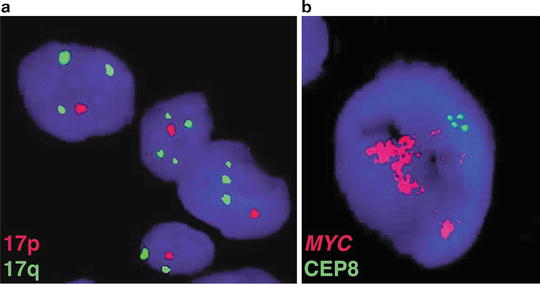
Fig. 9.2
Historical cytogenetic aberrations of medulloblastoma. (a) Interphase FISH performed on a medulloblastoma sample exhibiting copy number imbalance on chromosome 17, characterized by deletion of 17p and duplication of 17q (i[17]q). (b) A medulloblastoma sample with high-level amplification of the MYC proto-oncogene on chromosome 8q24 as demonstrated by FISH.
High-level amplification of the MYC proto-oncogene in the form of double-minute chromosomes has been observed for more than two decades, reported to occur in 5–10 % of patients (Fig. 9.2b). Historically, amplification of MYC has been recognized as a marker of poor patient outcome, often occurring in patients with a particularly aggressive form of the disease [3, 9–16].
Rare familial tumor syndromes, namely Gorlin and Turcot Syndromes, have provided considerable insight into the genetics underlying specific subsets of medulloblastoma patients [17]. Gorlin Syndrome (also referred to as Nevoid Basal Cell Carcinoma Syndrome, NBCCS) is an autosomal dominant disorder that results in abnormal facial and skeletal phenotypes, with affected individuals prone to the development of numerous basal cell carcinomas and predisposed to medulloblastoma. Germline loss-of-function mutations in the PTCH1 tumor suppressor gene on chromosome 9q are responsible for this disorder [18–20]. PTCH1 is a negative regulator of the Sonic Hedgehog (SHH) signal transduction pathway, an important developmental signaling cascade. As will be discussed in detail below, aberrant activation of the SHH pathway is found in ~25–30 % of all medulloblastoma patients and is perhaps the most well characterized molecular pathway involved in medulloblastoma development.
Turcot Syndrome is also an autosomal dominant condition; attributed to germline mutations in either APC or the DNA mismatch repair genes MLH1 or PMS2. Affected individuals are predisposed to either medulloblastoma (linked with APC mutation) or glioblastoma multiforme (linked with MLH1 or PMS2 mutations) [21]. The APC protein functions to control the activity of β-Catenin, the central molecule of the Wingless (WNT) signaling pathway. Activation of WNT signaling is observed in ~10–15 % of medulloblastomas, a distinct subset of patients with a highly favorable outcome, as will be discussed in detail below.
Molecular Subgroups of Medulloblastoma: Discovery and Initial Characterization
The Molecular Subgroup Concept
Clinical outcome of patients with medulloblastoma can be highly variable, irrespective of parameters routinely used in the clinic to predict patient risk (i.e., patient age, extent of resection, and metastatic stage). This is often exemplified by the very disparate therapeutic responses observed among patients with histologically identical disease. Furthermore, survival patterns reported for different age groups of medulloblastoma patients have strongly suggested underlying biological differences that must account for the distinct age of onset and therapeutic response of these patient subgroups [6, 22]. Much of this biological and clinical heterogeneity observed in medulloblastoma is now being explained by the recognition of unique molecular subgroups of the disease, a concept that has been supported for more than a decade now [23].
Hybridization of moderate-to-large series of RNAs isolated from primary medulloblastoma specimens to gene expression microarrays has revolutionized our concept of medulloblastoma as a disease. Based primarily on expression array profiling, numerous studies performed by independent laboratories have reported the existence of discrete molecular subgroups of medulloblastoma [24–28], each characterized by distinct genetics, cytogenetics and transcriptional profiles, as well as patient demographics, tumor phenotype, and clinical behavior (Figs. 9.3 and 9.4) [29]. In late 2010, a consensus meeting attended by leading groups in the medulloblastoma community was held where it was proposed that medulloblastoma should be regarded as consisting of a four-subgroup structure: WNT, SHH, Group 3, and Group 4 [30]. The impact of this consensus report and the studies that precipitated it have changed the way medulloblastoma is both viewed and studied in the basic research setting. Moreover, the implications for the development of clinical trials, revamping patient risk-stratification, and the administration of targeted therapy have been dramatic, all of which are now beginning to take molecular subgroup status into consideration.
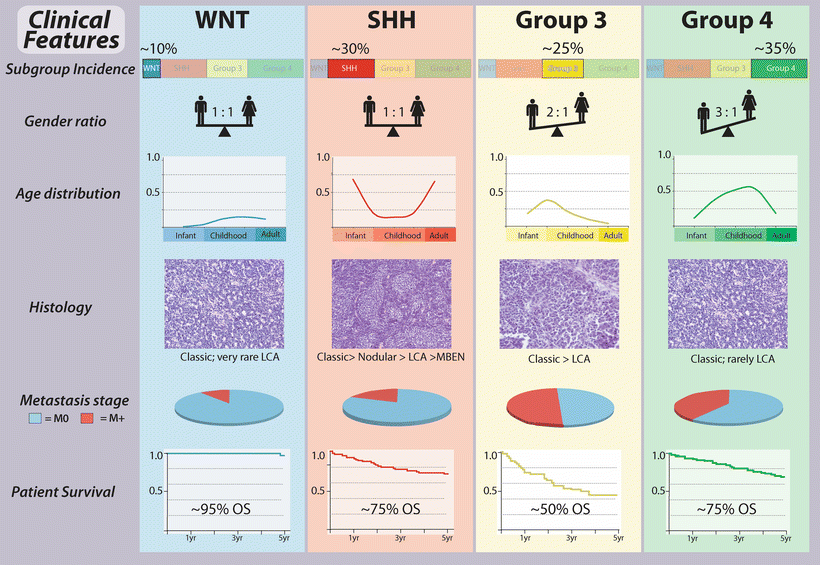
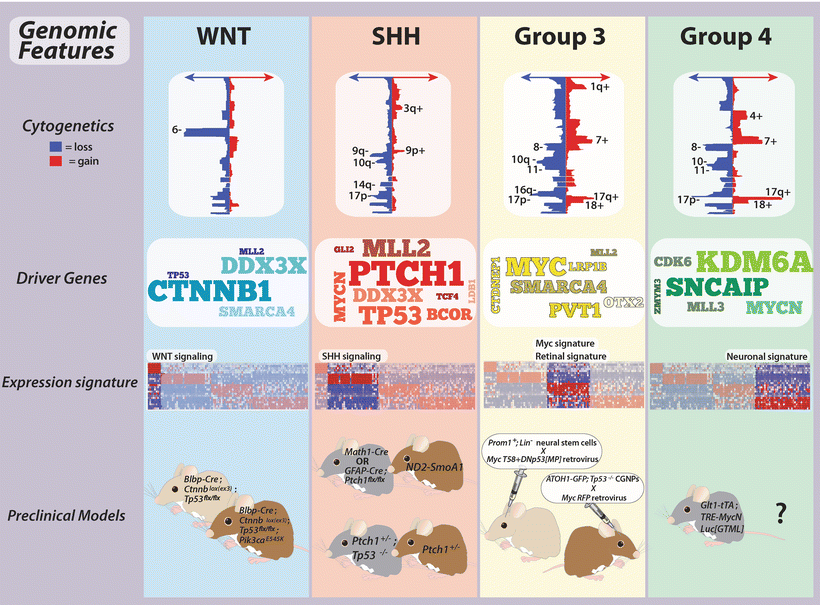
Fig. 9.3
Clinical features of medulloblastoma subgroups. General summary of the clinical characteristics intrinsic to the core molecular subgroups of medulloblastoma. LCA large cell and anaplastic, MBEN medulloblastoma with extensive nodularity, OS overall survival.
Fig. 9.4
Genomic features of medulloblastoma subgroups. Summary of the genomics of medulloblastoma subgroups, including cytogenetics, prominent driver genes, transcriptional signatures, and available preclinical models.
WNT Medulloblastoma
The least common of the four consensus medulloblastoma subgroups are those belonging to the WNT subgroup, accounting for just 10–15 % of cases. This subgroup affects a higher than expected proportion of female patients (male:female ratio of ~1:1 compared to an expected ratio of ~1.5:1 for all medulloblastomas) and is predominantly diagnosed in childhood and adolescence, almost never encountered in infants. WNT medulloblastomas are nearly without exception of classic histology and non-metastatic, currently having the best overall prognosis of any patient subgroup with cure rates of almost 100 % [4, 24, 27, 28, 31, 32].
Somatic missense mutations in exon 3 of CTNNB1 (which encodes β-Catenin) are present in >90 % of WNT medulloblastomas [33]. These mutations constitutively activate β-Catenin and prevent it from being degraded, resulting in its nuclear accumulation and consequent deregulation of WNT target genes. Monosomy 6 is another characteristic genetic feature of WNT subgroup medulloblastomas, highly enriched in this subgroup and found in the vast majority of cases [32, 34]. Immunohistochemistry (IHC) for β-Catenin and fluorescence in situ hybridization (FISH) for chromosome 6 constitute two commonly used “readouts” for WNT subgroup assignment, with either nucleo-positivity for β-Catenin or monosomy 6 acting as capable surrogates for identifying WNT subgroup patients (Fig. 9.5) [35]. More specific assays such as re-sequencing of CTNNB1 exon 3 performed in combination with additional state-of-the-art molecular profiling methods (discussed below) are now being recommended as more specific and sensitive methods for identifying this important subgroup of medulloblastoma patients.
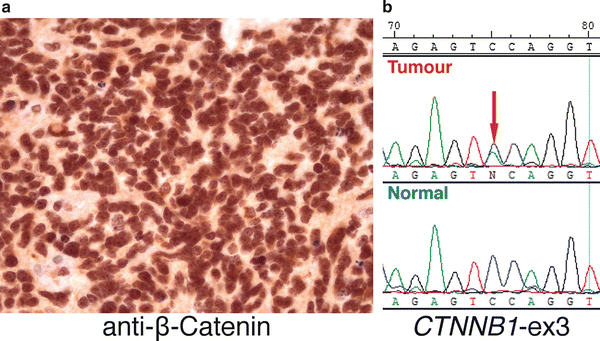
Fig. 9.5
Molecular assays for assignment of WNT medulloblastomas. (a) IHC shows strong nucleo-positivity typical of a WNT subgroup medulloblastoma. (b) DNA sequencing electropherogram showing a heterozygous CTNNB1 exon 3 mutation (red arrow) present in the tumor DNA of a WNT medulloblastoma (labeled “Tumor”) that is absent in matched normal germline control DNA (labeled “Normal”) from the same patient.
SHH Medulloblastoma
Medulloblastomas exhibiting aberrant activation of the SHH signaling pathway account for ~25–30 % of cases [32]. In infants (age 3 years or less) and older patients (age 16 years and up), SHH-activated cases predominate, accounting for up to 70 % of patients in these age groups [36]. The gender distribution for SHH subgroup medulloblastomas is comparable to what is seen among all medulloblastoma patients, with an observed male:female ratio of ~1.5:1. True desmoplastic and MBEN histologies appear to be largely restricted to this subgroup [35]; however, large cell/anaplastic (LCA) disease is also encountered, particularly in childhood cases. Metastatic disease (M+) is also observed but relatively uncommon. With respect to patient outcome, SHH cases appear to represent an intermediate prognosis class of patients [32], although specific patient subsets may have highly favorable or disparate clinical outcomes, depending on their underlying genotypes (as will be discussed later) [22, 37].
As was first verified in Gorlin Syndrome patients that develop medulloblastoma, PTCH1 is the prototypical tumor suppressor in the SHH subgroup, with somatic mutations inactivating PTCH1 confined to this subgroup. SUFU, another tumor suppressor functioning as a regulatory component of the SHH signaling pathway, is likewise mutated in this subgroup, both in the germline and somatically [38–40]. Loss of heterozygosity (LOH) on chromosomes 9q and 10q is enriched in SHH medulloblastomas [27, 34], presumably as a mechanism for inactivating the remaining wild-type allele in cases exhibiting PTCH1 or SUFU mutations, respectively. Similar to the use of β-Catenin nucleo-positivity as a biomarker for WNT medulloblastoma, comparable IHC-based methods have been proposed and implemented with modest success for the identification of SHH medulloblastomas, including SFRP1, GLI1, and GAB1, all of which exhibit apparent specificity for this subgroup [27, 28, 35, 41, 42].
Group 3 Medulloblastoma
The generically named Group 3 medulloblastomas account for ~25 % of all cases and appear to be restricted to pediatric patients, with only extremely rare instances reported in adults [42]. There is a male gender bias in Group 3, with an observed male:female ratio of ~2:1. M+ disease is common in Group 3 and has been documented in up to half of these cases. Group 3 subgroup affiliation currently carries with it the most dismal overall survival of the four subgroups, with only ~50 % of these patients or less alive at 5 years from the time of initial diagnosis [32].
Amplification of MYC is a characteristic oncogenic event observed in this subgroup, an event that is by and large restricted to Group 3 and found in 15–20 % of cases [34]. Cytogenetic aberrations such as gain of chromosomes 1q and 7, and loss of chromosomes 8p, 11, 16q and i[17]q are commonly observed in Group 3. Although preliminary biomarkers for Group 3 have been suggested (i.e., MYC amplification), there are currently no gold-standard single gene/marker assays for assigning medulloblastomas to this subgroup.
Group 4 Medulloblastoma
Group 4 medulloblastomas represent the most common patient subgroup, accounting for 35–40 % of all cases [32]. These tumors occur across all age groups but constitute the most predominant form of the disease in childhood and adolescence. There is a strong gender bias in Group 4, with an observed male:female ratio of ~3:1. Metastatic disease is observed in approximately one-third of Group 4 patients. Similar to the SHH subgroup, Group 4 patients tend to comprise an intermediate outcome subgroup [32], although there is growing evidence for clinical heterogeneity within this large fraction of patients [43].
Cytogenetically, Group 4 medulloblastomas share some commonalities with Group 3, most notably being the highly prevalent i[17]q which is noted in up to 70–80 % of Group 4’s. Chromosome X loss in female Group 4 patients is also a frequent occurrence. Compared to other subgroups, Group 4 medulloblastomas remain the least well understood with regards to oncogenic driver genes, although a few interesting novel candidates have emerged from recent genomic studies, as will be discussed in detail below.
Next-Generation Genomics of Medulloblastoma: Assigning Driver Genes to Medulloblastoma Subgroups
Recent technological breakthroughs in the field of genomics have dramatically improved the resolution at which the cancer genome is studied [44, 45]. Application of high-density microarray platforms and next-generation sequencing (NGS) to medulloblastoma has led to the identification of a host of novel candidate genes that are recurrently affected by somatic copy number alterations (SCNAs) or mutation, and sometimes both (Figs. 9.4, 9.6, and 9.7) [33]. Many of these events appear to be enriched or restricted to a particular subgroup and are thus likely playing an integral role in the biology driving the initiation, maintenance, and progression of the subgroup(s) harboring the mutational event. Genes and pathways emerging as important oncogenic drivers in medulloblastoma, including how they are distributed within the subgroups, are described below.
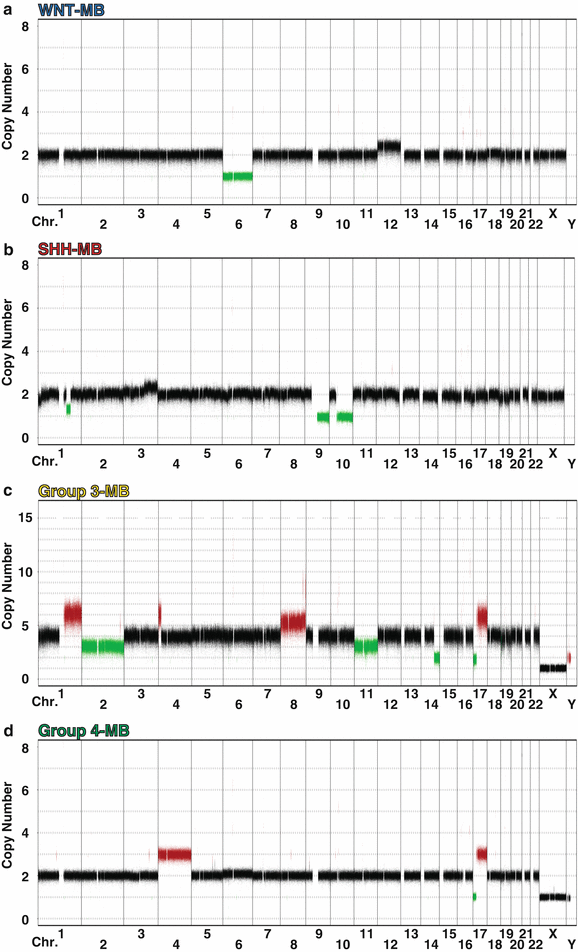
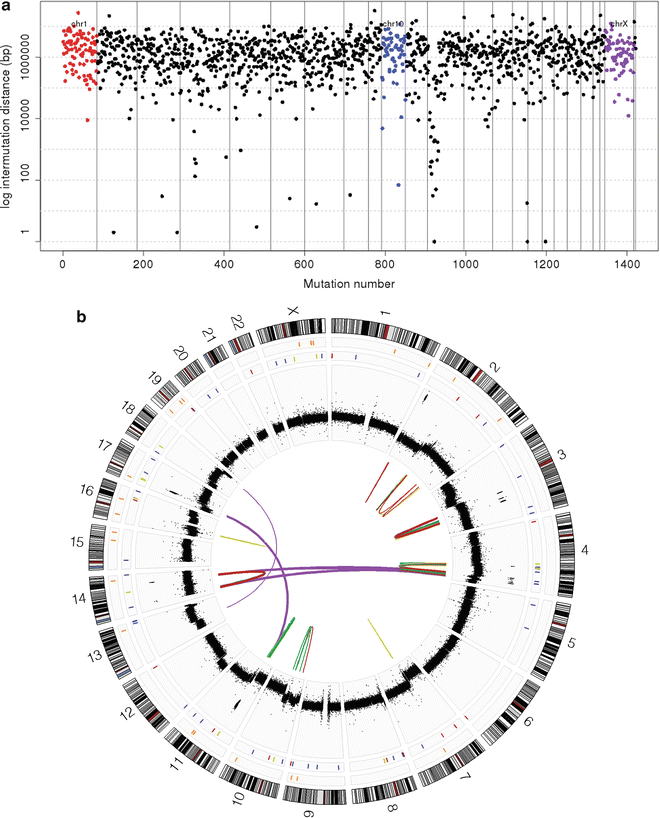
Fig. 9.6
Characteristic cytogenetics of medulloblastoma subgroups. Genome-wide copy number profiles highlighting chromosomal gains and losses typical of each of the four subgroups. (a) A WNT subgroup medulloblastoma exhibiting monosomy 6 and an otherwise balanced genome. (b) A SHH subgroup medulloblastoma characterized by signature deletions of chromosomes 9q and 10q. (c) A Group 3 medulloblastoma with prototypical gains of chromosome 1q and chromosome 8 (including MYC amplification), as well as an isochromosome 17q (i[17]q). (d) A Group 4 medulloblastoma exhibiting gain of chromosome 4 and i[17]q.
Fig. 9.7
Next-generation sequencing of medulloblastoma. (a) Rainfall plot depicting the distribution of somatic mutations (single nucleotide variants; SNVs) across the genome of a SHH subgroup medulloblastoma as determined by NGS. (b) Circos plot of NGS data for a SHH subgroup medulloblastoma exhibiting excessive genomic rearrangements and somatic copy number alterations (SCNAs).
Known Cancer Genes
A number of genes previously implicated in medulloblastoma have now been accurately placed in the context of the molecular subgroups. Several of these candidates were once thought to be rarely affected when studying medulloblastoma as a single entity, but are now considered of higher relevance given their subgroup-specificity and increased frequency within a particular subgroup.
The TP53 tumor suppressor, classically reported as being somatically mutated in only ~5 % of medulloblastomas, has now been observed to be almost exclusively mutated in WNT and SHH subgroup cases, affecting ~10–15 % of cases from each subgroup [37, 46]. Moreover, germline mutations in TP53, the hallmark genetic event causing Li-Fraumeni Syndrome (LFS) [47, 48], a condition predisposing to the development of a variety of different cancers including medulloblastoma, have now been confirmed to be restricted to the SHH subgroup [49], especially in childhood and adolescent patients.
In contrast to the MYC proto-oncogene which is amplified exclusively in Group 3, high-level amplifications of MYCN are found in both SHH and Group 4 but rarely in Group 3 and never in WNT medulloblastomas [34, 43]. Likewise, copy number gains of OTX2, a developmental transcription factor previously implicated in medulloblastoma pathogenesis [50–54], appear to be restricted to Groups 3 and 4 [34, 55], suggesting it plays an important role in the biology of these tumors.
Additional oncogenic copy number alterations showing enrichment in SHH-driven medulloblastoma now include (but are not limited to) amplification of GLI2, MYCL1, PPM1D, YAP1, IGF1R, IRS2, MDM4, and miR–17/92 and focal homozygous deletion of PTEN and PTCH1 [34, 56, 57]. These events collectively suggest that at least three main pathways contribute to the majority of SHH-driven medulloblastomas: SHH signaling, RTK/PI3K signaling, and TP53 signaling [34].
DDX3X
Identified as a common target of recurrent mutation in three parallel NGS studies of medulloblastoma [58–60], DDX3X is among the newest candidates to be implicated as an important medulloblastoma driver gene. DDX3X is a DEAD-box RNA helicase that has been shown to play a role in a variety of cellular processes, ranging from chromosome segregation to transcription and translation [61–64]. Mutations in DDX3X are confined to either of its two helicase domains and are always non-truncating variants [33], suggesting that these mutations alter the function of DDX3X rather than causing loss-of-function [59, 60]. Approximately half of all WNT cases harbor a DDX3X mutation, whereas 10–15 % of SHH cases are likewise mutated. Recent NGS of adult SHH medulloblastomas has revealed a high proportion of DDX3X SNVs, suggesting this candidate is particularly important in the biology of adult SHH cases. In contrast, mutations in DDX3X are seldomly observed in Groups 3 and 4.
Chromatin Modifiers
One of the most unexpected findings disclosed from recent genomic studies of medulloblastoma concerns the high frequency of mutations and copy number alterations affecting chromatin modifiers [33, 65, 66]. In 2009, Northcott and colleagues reported a series of infrequent but recurrent SCNAs targeting histone lysine methyltransferases, histone lysine demethylases, histone acetyltransferases, and chromatin remodelers [67]. A subsequent landmark exon re-sequencing study performed by Parsons et al. identified recurrent and mutually exclusive mutations in histone 3, lysine 4 (H3K4) methyltransferases, MLL2 and MLL3, collectively mutated in ~16 % of surveyed cases [68]. Since these two initial reports implicating deregulation of the chromatin machinery in medulloblastoma, this theme has been further substantiated in all recent medulloblastoma NGS studies, now in the context of molecular subgroups [33, 58–60]. Interestingly, MLL2 mutations have been confirmed to be more common in WNT and SHH tumors, whereas MLL3 mutations are more prevalent in Groups 3 and 4. The SWI/SNF family gene SMARCA4 that encodes BRG1, a component of a multi-protein chromatin-remodeling complex, is recurrently inactivated in WNT and Group 3 medulloblastomas. Similarly, chromatin-modifying genes LDB1, BCOR, and LMO4 are targeted either by mutations, copy number alterations, or both exclusively in SHH-driven cases. Finally, KDM6A, a histone 3, lysine 27 (H3K27) demethylase, appears to be inactivated by either somatic mutation or focal deletion specifically in Group 4. Moreover, EZH2, which imposes the opposite function of KDM6A, catalyzing the trimethylation of H3K27 (H3K27me3) is aberrantly over-expressed in Groups 3 and 4, suggesting a propensity for an aberrant H3K27 methylation state in these subgroups [60, 69]. Collectively, this series of recent observations strongly supports the notion that deregulation of the histone code is a key event in medulloblastoma pathogenesis, with somatic alterations occurring in a high proportion of cases across the four subgroups.
Atypical Structural Variation in Medulloblastoma
In addition to the spectrum of genes described above as recurrently mutated or affected by SCNAs, more complex mechanisms of deregulation, including both gene-specific and genome-wide structural rearrangements have been uncovered during the genomics era of medulloblastoma (Fig. 9.8). These recurrent structural variants appear to be common to medulloblastoma and may play an even bigger role than standard SNVs and SCNAs affecting protein-coding sequences.
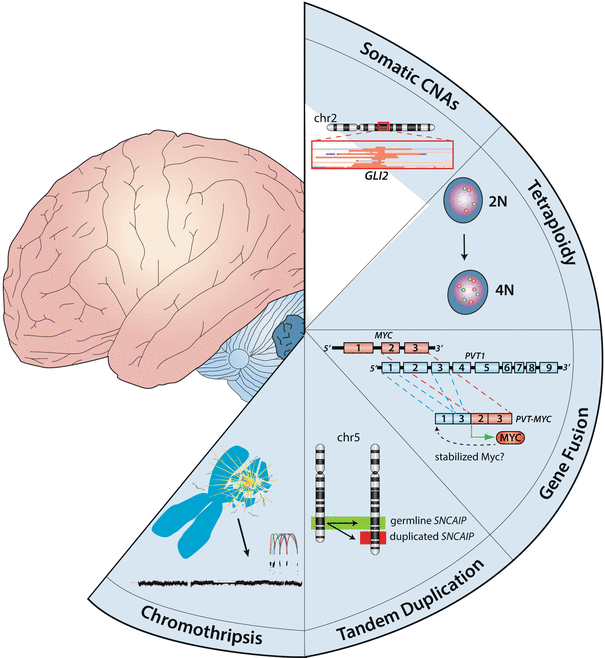
Fig. 9.8
Mechanisms of recurrent structural variation in medulloblastoma. Cartoon showing the different mechanisms responsible for the prominent structural variation reported in medulloblastoma. Depicted types of structural variation include SCNAs, tetraploidy, gene fusions, tandem duplications, and chromothripsis.
NGS of a series of LFS medulloblastomas recently identified massive chromosomal rearrangements known as chromothripsis (Fig. 9.7b) [49]—excessive genomic rearrangements (i.e., inversions, amplifications, deletions) clustered on one or a few chromosomes presumed to have arisen from a single catastrophic DNA breakage/repair event during tumorigenesis [70–72]. This phenomenon was observed in the majority of LFS medulloblastomas analyzed, was specifically enriched in SHH medulloblastoma, and co-occurred with cases harboring either germline or somatic TP53 mutations [49]. Importantly, chromothripsis observed in these cases often resulted in amplification of known medulloblastoma oncogenes (i.e., MYCN and GLI2), providing a mechanism for the activation of these critical driver genes.
Using a combination of single nucleotide polymorphism (SNP) arrays and RNA sequencing (RNASeq), Northcott et al. reported PVT1–MYC fusion genes as being highly recurrent and specific to MYC-amplified Group 3 medulloblastoma [34]. This is the first example of a recurrent fusion gene identified in medulloblastoma and is suspected to potentiate MYC activity by yet an unknown mechanism. In the same study, tandem duplications affecting the SNCAIP gene on chromosome 5q were reported in up to ~25 % of a particular Group 4 subtype. SNCAIP is a neuronal gene implicated in Parkinson’s disease [73, 74]; how these alterations contribute to Group 4 medulloblastoma biology currently remains unclear.
Whole genome duplication (i.e., tetraploidy) has been observed in medulloblastoma karyotypes for more than two decades [75, 76]. Now as a result of NGS, tumor cell ploidy can be readily estimated based on allele frequencies of SNPs and SNVs present in the genome. In a recent report by Jones et al., up to one-third of investigated medulloblastoma genomes were shown to be tetraploid, with higher frequencies noted in Groups 3 and 4 [58]. The significance of the high frequency of tetraploidy observed in medulloblastoma, particularly in Groups 3 and 4, will require further investigation but is thought to contribute to the overall genomic instability (i.e., gains and losses of whole chromosomes or chromosome arms) often noted in these tumors.
Preclinical Models of Medulloblastoma: Validating the Genetics of the Human Disease
Understanding the biological consequences of the genetic and epigenetic events observed in medulloblastoma and its subgroups requires accurate and faithful preclinical models that allow for comprehensive in vitro and in vivo functional studies. A series of established, immortalized medulloblastoma cell lines derived from human patient samples have been in use for the past 20–30 years [77–81]. These models have served as convenient systems for a variety of purposes including, evaluating candidate gene function, investigating genetic, epigenetic, and transcriptional alterations, and testing novel therapies, all in the context of medulloblastoma. Now with the recognition of medulloblastoma subgroups and increasing knowledge of the genetics underlying these subgroups, the validity of these “workhorse” medulloblastoma models has been put into question. Ongoing genomic analysis of these lines suggests they do not faithfully recapitulate the four medulloblastoma subgroups and harbor events not observed in primary medulloblastoma counterparts as a result of their continued evolution during long-term passage in culture [34]. Novel, low-passage lines and medulloblastoma xenograft models that require passaging in the mouse have recently emerged as possible solutions to the caveats associated with immortalized, high-passage cell lines and will likely be more heavily relied upon in the future [82, 83].
An immense amount of knowledge regarding the developmental biology of medulloblastoma has been gained from the use of genetically engineered mouse models [84]. The majority of such murine models generated and studied to date have been driven by activation of the SHH pathway in neuronal progenitor and stem cell populations [85]. Germline inactivation of one copy of the Ptch1 gene (often referred to as Ptc +/− mice) results in 15–20 % of mice developing cerebellar tumors that are histologically similar to human medulloblastomas and exhibit aberrant SHH pathway activation suggesting they are accurate models of this subgroup [86, 87]. Combining loss of Ptch1 with inactivation of Trp53 (i.e., Ptc +/− ; Trp53 −/−) dramatically increases tumor incidence and reduces latency, with up to ~95 % of mice developing medulloblastoma within 12 weeks [88]. Several other SHH-activated mouse models have been generated, including those driven by an activated Smoothened transgene [89–91], homozygous germline deletion of Ptch1 in specific cell types [92], those driven by administration of SHH ligand with cooperating oncogenes [93–95], and others [96–99]. These models have led to a better understanding of the genetics underlying SHH-driven medulloblastoma, the probable cells-of-origin for this subgroup, and provided the research community with tools for asking an array of questions related to medulloblastoma biology.
Representative models for the remaining subgroups have also been published, providing important clues regarding their differing biologies. Gibson et al. expressed a constitutively active form of β-Catenin (i.e., Ctnnb1 Δex3) in progenitor cells of the developing hindbrain, successfully generating the first WNT-driven medulloblastoma mouse model [100]. More recently, complementary studies by Pei et al. and Kawauchi et al. combined over-expression of Myc with loss of wild-type Trp53 in orthotopic transplantation models to generate tumors resembling human Group 3 medulloblastoma [101, 102]. Finally, a model relying on transgenic over-expression of Mycn is believed to represent the lone Group 4 medulloblastoma model currently available [103].
As a plethora of new candidate medulloblastoma genes have recently been discovered, it is anticipated that many novel models based on these genes and rational gene combinations will be generated and introduced during the next few years. These models will serve not only to functionally validate events observed in the human disease but also to further progress our understanding of their role in disease biology and evaluate their relevance and utility as potential targets for molecularly informed therapy.
Translational Significance of the Medulloblastoma Genomics Era
Molecular Classification and Risk-Stratification
Medulloblastoma subgroups exhibit highly disparate molecular genetics and clinical characteristics, suggesting they should be treated as different diseases in the clinic. Before such a concept is put into general practice, robust, highly accurate, and efficient methods that are accessible to treating physicians for establishing subgroup assignments are necessary. Novel assays that are gaining an appreciation in this arena include the use of DNA methylation arrays and platforms for measuring the expression of custom gene panels. Both Schwalbe et al. and Hovestadt et al. have recently demonstrated that DNA methylation arrays can be used to assign medulloblastoma subgroups with high confidence, including samples derived from formalin-fixed paraffin embedded (FFPE) material [104, 105]. RNA-based methods such as the nanoString assay have also shown utility at subgrouping of samples preserved in FFPE [106] (Fig. 9.9). Further validation of these methods and potentially others in the setting of medulloblastoma clinical trials is expected in the near future as the interest to subgroup patients in a prospective manner increases.
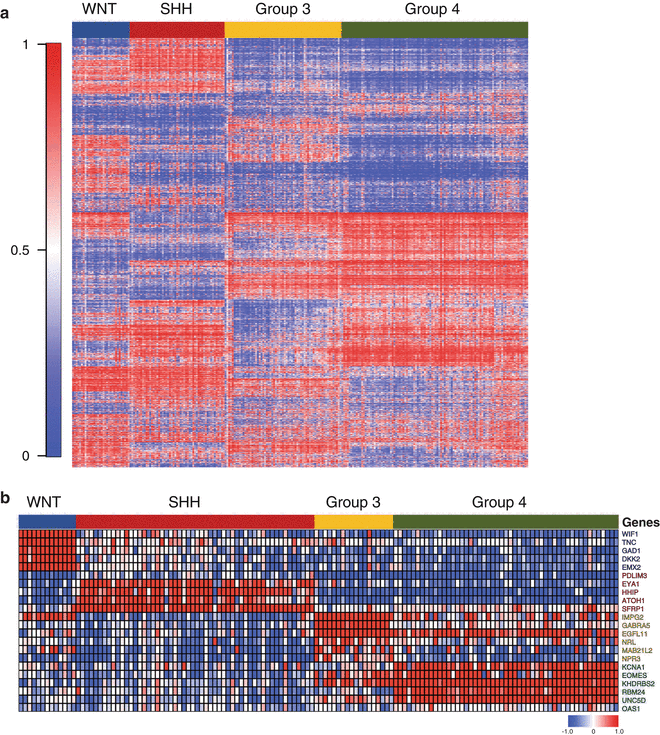
Fig. 9.9
Conventional assays for the molecular classification of medulloblastoma. (a) Heatmap of DNA methylation array (Illumina 450K platform) data for >250 primary medulloblastomas classified according to their appropriate molecular subgroup. Data was generated using DNAs extracted from either fresh-frozen tumor tissue or FFPE material. (b) Heatmap of gene expression data derived from a custom nanoString assay consisting of 22-signature genes for >100 medulloblastomas classified by molecular subgroup. Data was generated using RNAs extracted from FFPE material.
In light of the excellent prognosis associated with WNT medulloblastoma patients, plans to de-escalate craniospinal radiation or even eliminate it in these patients will be implemented in forthcoming clinical trials. Similarly, prospective stratification of all Group 3 patients into a high-risk treatment category is likewise being considered.
Another subset of patients that appears to be of significant clinical relevance are those with TP53-mutated SHH medulloblastomas. Using large retrospective patient cohorts, Zhukova et al. demonstrated that the dismal outcome sometimes attributed to medulloblastomas harboring TP53 mutation [107] can be explained by considering patient subgroup information [37]. TP53-mutated cases within the SHH subgroup exhibit a significantly worse outcome compared to either subgroup-matched non-mutated counterparts or WNT cases likewise harboring TP53 mutation. Furthermore, the prevalence of chromothripsis and oncogene amplification observed in TP53-mutated SHHs suggest these patients should be stratified as a unique risk-group and possibly subjected to treatments tailored for their genotype.
Targeting Medulloblastoma with Rational Therapies
One of the major goals motivating the comprehensive genomic characterization of medulloblastoma is the identification of targets that can be specifically exploited for future treatment of the disease. To date, antagonists of the SHH pathway, acting mainly at the level of SMO, have demonstrated the most promise [108–112]. Compounds such as GDC-0449 from Genentech have shown dramatic although transient tumor regression when administered to patients with metastatic medulloblastoma, with patients eventually becoming resistant to the targeted therapy [111, 113]. Similar acquired resistance has been noted when related SMO antagonists (i.e., LDE-225) have been used to treat mouse models of the disease [110]. Ongoing efforts aim to combine SHH antagonists with additional inhibitory agents targeting cooperating pathways in hopes of achieving an improved and more sustained response to treatment. Furthermore, genomic analysis of human SHH medulloblastomas suggests that not all SHH-driven cases are likely to respond to inhibitors acting at the level of SMO, as subsets of cases such as those exhibiting amplification of downstream pathway components (i.e., GLI2) are likely to have primary resistance to these agents [34, 67]. As such, screening patients for both SHH subgroup affiliation and their mutation/copy number status in select SHH pathway genes prior to treatment with the current generation of SHH pathway inhibitors could improve their likelihood of response in the future. In addition, novel approaches targeting pathway components downstream of SMO, including the use of agents inhibiting GLI family transcription factors, are currently being evaluated and may increase the likelihood of response when combined with conventional SHH antagonists [114, 115].
The frequent deregulation of chromatin modifiers in medulloblastoma and the potential consequences of these events on the underlying epigenome make the prospect of epigenetic therapy an attractive possibility for medulloblastoma patients [65, 66]. Histone deacetylase (HDAC) inhibitors such as Vorinostat are currently being evaluated in medulloblastoma clinical trials [116, 117]. Similarly, 3-deazaneplanocin A (DZNep), a potent inhibitor of EZH2, is now being prioritized as an agent to be tested in upcoming clinical trials for medulloblastoma [118]. Similar agents targeting histone-modifying enzymes are presently being evaluated in the research setting and undoubtedly will enter the clinical trials arena for medulloblastoma patients in the near future.
Summary
Considerable advances have recently been made with respect to our understanding of the molecular genetics underlying medulloblastoma (Fig. 9.10). Acknowledgement of unique molecular subgroups and an improved knowledge of the genes and pathways responsible for their pathogenesis can now at least partially explain the long-recognized biological and clinical heterogeneity encountered in the disease. Application of next-generation genomic platforms to large patient cohorts has identified new driver genes recurrently mutated in specific medulloblastoma subgroups, in addition to recurrent and often complex structural rearrangements, all at base-pair resolution. Additionally, new assays for rapidly confirming subgroup affiliation with pinpoint accuracy have been developed and are now making their way into clinical trials, as the need to progressively subgroup patients in the clinical setting intensifies. As the medulloblastoma research community harnesses the wealth of information gained during the current medulloblastoma “genomics era,” new preclinical models faithfully recapitulating the genetics and the biology of the human subgroups are emerging. These new models will serve as valuable tools for the identification, development, and evaluation of rational therapies, bridging the gap between discoveries made in the research laboratory and the future administration of more specific, less-toxic targeted treatment options. It can be anticipated that we are on the verge of an era of personalized medicine for medulloblastoma, whereby patients will be treated with therapies specifically tailored to their underlying genotypes. It is hoped that these novel diagnostic and therapeutic strategies will improve outcomes and quality of life for medulloblastoma patients going forward.
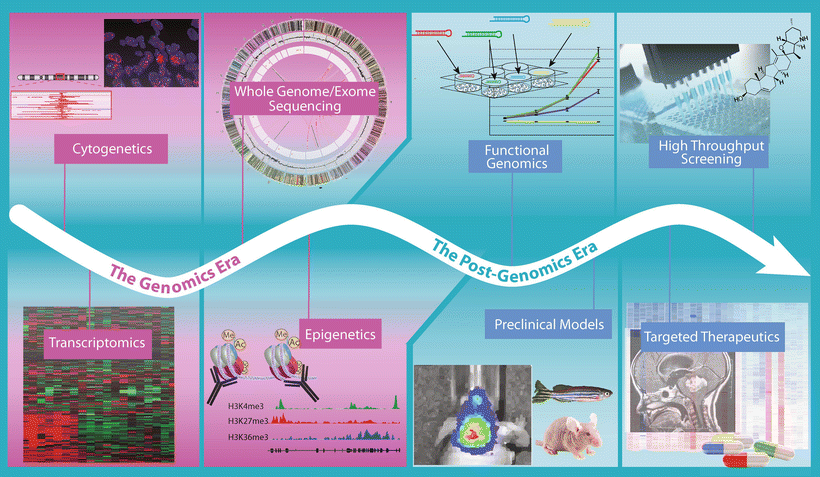
Fig. 9.10
Progression from genomics to the post-genomic era of medulloblastoma. Timeline highlighting the technological approaches being applied in medulloblastoma studies during the current “Genomics Era” and those that will become the focus of the imminent “Post-Genomics Era”.
CNS-PNET
Central nervous system primitive neuroectodermal tumor (CNS-PNET, also known as supratentorial PNET to distinguish them from medulloblastoma) is a catch-all term for what is an extremely heterogeneous group of tumors arising in the cerebrum or spinal cord. They are one of the least clearly demarcated entities in terms of their histology, comprising a wide morphological spectrum. The WHO defines them as “An embryonal tumor composed of undifferentiated or poorly differentiated neuroepithelial cells which have the capacity for, or display, divergent differentiation along neuronal, astrocytic, muscular or melanocytic lines” [1]. Four variants of CNS-PNET are described in addition to “not otherwise specified” (NOS), namely: CNS neuroblastoma, CNS ganglioneuroblastoma, medulloepithelioma, and ependymoblastoma (EBL). CNS neuroblastoma is reserved for primitive tumors with solely neuronal differentiation, while ganglioneuroblastoma additionally implies the presence of ganglion cells. A further PNET occurring throughout the brain (including infratentorial locations) and characterized by broad bands of neuropil with true rosettes surrounding lumens, known as embryonal tumor with abundant neuropil and true rosettes (ETANTR), has also been noted but is not yet included in the WHO classification [119]. All of these variants are considered to be of malignancy Grade IV [1].
There is no clear shift in the gender distribution of CNS-PNETs, with a male:female ratio of ~1.2:1. The majority of these tumors occur in children, with a mean age at diagnosis of ~5.5 years [1]. Rarer cases in adults have also been reported, but with an incidence rate approximately one-fifth of that in children [120]. CNS-PNETs diagnosed in older patients are associated with a significantly worse prognosis than those occurring before the age of 40, and also with a worse outcome than for adult medulloblastoma (median survival 16 months for adult CNS-PNET vs. 155 months for medulloblastoma [121]). The latter trend holds true for pediatric cases, with 5-year overall survival typically below 50 % for CNS-PNET compared with 70–80 % for medulloblastoma [122, 123]. Pineal tumors in particular seem to be associated with poor outcomes. Treatment options are also less well defined than those for medulloblastoma, with some evidence that CNS-PNETs are resistant to standard Packer chemotherapy regimens [124], and no clear rationale for molecularly targeted therapies has been proposed. This is especially true for infants, who do not receive craniospinal radiation due to the risk of severe developmental defects. Slightly better outcomes have been reported, however, in patients receiving risk-adapted radiotherapy followed by high-dose cyclophosphamide-based chemotherapy and stem cell rescue [125]. As with medulloblastoma, survivors often experience significant morbidities and a reduction in quality of life due to tumor- and treatment-related sequelae.
Somatic Copy Number Alterations
Several studies have used methods of varying resolution to investigate SCNAs in CNS-PNET over the last 10–15 years. The general picture that has emerged is that these tumors do not typically display any of the common changes seen in medulloblastoma, such as i[17q], but may harbor other changes. Recently, a region of focal amplification on chromosome 19q has been identified as a highly recurrent alteration in certain subsets of CNS-PNET, as will be discussed in more detail in a later section.
One of the earliest studies comparing the cytogenetics of CNS-PNETs to medulloblastoma, for example, noted a higher frequency of chromosome 14q and 19q loss in supratentorial compared with infratentorial tumors, and no 17q gain in the supratentorial cases [126]. Another early report described an amplification of the TERT gene in a recurrence of a medulloepithelioma, which was not seen in the primary tumor, suggesting a role in tumor progression [127]. The authors also described increased expression of telomerase as a common finding in CNS-PNET. A lack of 17q gain but recurrent loss of 13q was noted in an array-based study of CNS-PNETs, which also reported amplifications of PDGFRA/KIT, MYB and 19q, as well as homozygous CDKN2A/B deletion [128]. One subsequent report did identify chromosome 17 alterations in 2/10 CNS-PNETs, but again noted that this change was significantly less frequent than in medulloblastoma [129]. The same study reported regions of loss on 1p and gain of 19p as being recurrent in CNS-PNET, and described deletions of CDKN2A/B in 7/21 cases examined [129]. Recurrent gain of 19p was confirmed in a more recent study of 29 CNS-PNETs, along with gains on 2p and 1q, which were seen in more than 20 % of cases [130]. Focal loss of CADPS on 3p was observed in 28 % of samples, and tumors showing this loss carried a worse prognosis [130]. In addition to CDKN2A/B deletions, SCNAs at other key cell-cycle regulatory genes seem to be a relatively common event in CNS-PNET, with 5/20 cases in one study displaying focal amplifications of CDK4, CDK6, CCND1, or CCND2 [131].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


