5 | Principles of Clinical Pharmacology |
Drugs are the cornerstone of modern therapeutics. Nevertheless, it is well recognized among physicians and in the lay community that the outcome of drug therapy varies widely among individuals. While this variability has been perceived as an unpredictable, and therefore inevitable, accompaniment of drug therapy, this is not the case. The goal of this chapter is to describe the principles of clinical pharmacology that can be used for the safe and optimal use of available and new drugs.
Drugs interact with specific target molecules to produce their beneficial and adverse effects. The chain of events between administration of a drug and production of these effects in the body can be divided into two components, both of which contribute to variability in drug actions. The first component comprises the processes that determine drug delivery to, and removal from, molecular targets. The resulting description of the relationship between drug concentration and time is termed pharmacokinetics. The second component of variability in drug action comprises the processes that determine variability in drug actions despite equivalent drug delivery to effector drug sites. This description of the relationship between drug concentration and effect is termed pharmacodynamics. As discussed further below, pharmacodynamic variability can arise as a result of variability in function of the target molecule itself or of variability in the broad biologic context in which the drug-target interaction occurs to achieve drug effects.
Two important goals of the discipline of clinical pharmacology are (1) to provide a description of conditions under which drug actions vary among human subjects; and (2) to determine mechanisms underlying this variability, with the goal of improving therapy with available drugs as well as pointing to new drug mechanisms that may be effective in the treatment of human disease. The first steps in the discipline were empirical descriptions of the influence of disease on drug actions and of individuals or families with unusual sensitivities to adverse drug effects. These important descriptive findings are now being replaced by an understanding of the molecular mechanisms underlying variability in drug actions. Thus, the effects of disease, drug coadministration, or familial factors in modulating drug action can now be reinterpreted as variability in expression or function of specific genes whose products determine pharmacokinetics and pharmacodynamics. Nevertheless, it is often the personal interaction of the patient with the physician or other health care provider that first identifies unusual variability in drug actions; maintained alertness to unusual drug responses continues to be a key component of improving drug safety.
Unusual drug responses, segregating in families, have been recognized for decades and initially defined the field of pharmacogenetics. Now, with an increasing appreciation of common and rare polymorphisms across the human genome, comes the opportunity to reinterpret descriptive mechanisms of variability in drug action as a consequence of specific DNA variants, or sets of variants, among individuals. This approach defines the field of pharmacogenomics, which may hold the opportunity of allowing practitioners to integrate a molecular understanding of the basis of disease with an individual’s genomic makeup to prescribe personalized, highly effective, and safe therapies.
IDENTIFYING DRUG TARGETS
Drug therapy is an ancient feature of human culture. The first treatments were plant extracts discovered empirically to be effective for indications like fever, pain, or breathlessness. This symptom-based empiric approach to drug development was supplanted in the twentieth century by identification of compounds targeting more fundamental biologic processes such as bacterial growth or elevated blood pressure; the term “magic bullet,” coined by Paul Ehrlich to describe the search for effective compounds for syphilis, captures the essence of the hope that understanding basic biologic processes will lead to highly effective new therapies. An integral step in modern drug development follows identification of a chemical lead with biologic activity with increasingly sophisticated medicinal chemistry-based structural modifications to develop compounds with specificity for the chosen target, lack of “off-target” effects, and pharmacokinetic properties suitable for human use (e.g., consistent bioavailability, long elimination half-life, no high-risk pharmacokinetic features described further below).
A common starting point for contemporary drug development is basic biologic discovery that implicates potential target molecules: examples of such target molecules include HMG-CoA reductase or the BRAF V600E mutation in many malignant melanomas. The development of compounds targeting these molecules has not only revolutionized treatment for diseases such as hypercholesterolemia or malignant melanoma, but has also revealed new biologic features of disease. Thus, for example, initial spectacular successes with vemurafenib (which targets BRAF V600E) were followed by near-universal tumor relapse, strongly suggesting that inhibition of this pathway alone would be insufficient for tumor control. This reasoning, in turn, supports a view that many complex diseases will not lend themselves to cure by targeting a single magic bullet, but rather single drugs or combinations will need to attack multiple pathways whose perturbation results in disease. The use of combination therapy in settings such as hypertension, tuberculosis, HIV infection, and many cancers highlights potential for such a “systems biology” view of drug therapy.
GLOBAL CONSIDERATIONS
![]() It is true across all cultures and diseases that factors such as compliance, genetic variants affecting pharmacokinetics or pharmacodynamics, and drug interactions contribute to drug responses. In addition, culture- or ancestry-specific factors play a role. For example, the frequency of specific genetic variants modulating drug responses often varies by ancestry, as discussed later. Cost issues or cultural factors may determine the likelihood that specific drugs, drug combinations, or over-the-counter (OTC) remedies are prescribed. The broad principles of clinical pharmacology enunciated here can be used to analyze the mechanisms underlying successful or unsuccessful therapy with any drug.
It is true across all cultures and diseases that factors such as compliance, genetic variants affecting pharmacokinetics or pharmacodynamics, and drug interactions contribute to drug responses. In addition, culture- or ancestry-specific factors play a role. For example, the frequency of specific genetic variants modulating drug responses often varies by ancestry, as discussed later. Cost issues or cultural factors may determine the likelihood that specific drugs, drug combinations, or over-the-counter (OTC) remedies are prescribed. The broad principles of clinical pharmacology enunciated here can be used to analyze the mechanisms underlying successful or unsuccessful therapy with any drug.
INDICATIONS FOR DRUG THERAPY: RISK VERSUS BENEFIT
It is self-evident that the benefits of drug therapy should outweigh the risks. Benefits fall into two broad categories: those designed to alleviate a symptom and those designed to prolong useful life. An increasing emphasis on the principles of evidence-based medicine and techniques such as large clinical trials and meta-analyses have defined benefits of drug therapy in broad patient populations. Establishing the balance between risk and benefit is not always simple. An increasing body of evidence supports the idea, with which practitioners are very familiar, that individual patients may display responses that are not expected from large population studies and often have comorbidities that typically exclude them from large clinical trials. In addition, therapies that provide symptomatic benefits but shorten life may be entertained in patients with serious and highly symptomatic diseases such as heart failure or cancer. These considerations illustrate the continuing, highly personal nature of the relationship between the prescriber and the patient.
Adverse Effects Some adverse effects are so common and so readily associated with drug therapy that they are identified very early during clinical use of a drug. By contrast, serious adverse effects may be sufficiently uncommon that they escape detection for many years after a drug begins to be widely used. The issue of how to identify rare but serious adverse effects (that can profoundly affect the benefit-risk perception in an individual patient) has not been satisfactorily resolved. Potential approaches range from an increased understanding of the molecular and genetic basis of variability in drug actions to expanded postmarketing surveillance mechanisms. None of these have been completely effective, so practitioners must be continuously vigilant to the possibility that unusual symptoms may be related to specific drugs, or combinations of drugs, that their patients receive.
Therapeutic Index Beneficial and adverse reactions to drug therapy can be described by a series of dose-response relations (Fig. 5-1). Well-tolerated drugs demonstrate a wide margin, termed the therapeutic ratio, therapeutic index, or therapeutic window, between the doses required to produce a therapeutic effect and those producing toxicity. In cases where there is a similar relationship between plasma drug concentration and effects, monitoring plasma concentrations can be a highly effective aid in managing drug therapy by enabling concentrations to be maintained above the minimum required to produce an effect and below the concentration range likely to produce toxicity. Such monitoring has been widely used to guide therapy with specific agents, such as certain antiarrhythmics, anticonvulsants, and antibiotics. Many of the principles in clinical pharmacology and examples outlined below, which can be applied broadly to therapeutics, have been developed in these arenas.
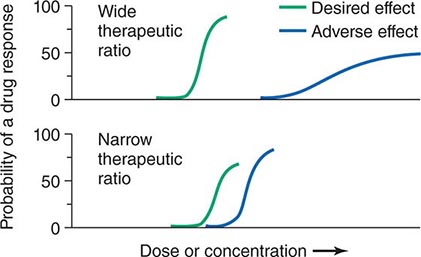
FIGURE 5-1 The concept of a therapeutic ratio. Each panel illustrates the relationship between increasing dose and cumulative probability of a desired or adverse drug effect. Top. A drug with a wide therapeutic ratio, i.e., a wide separation of the two curves. Bottom. A drug with a narrow therapeutic ratio; here, the likelihood of adverse effects at therapeutic doses is increased because the curves are not well separated. Further, a steep dose-response curve for adverse effects is especially undesirable, as it implies that even small dosage increments may sharply increase the likelihood of toxicity. When there is a definable relationship between drug concentration (usually measured in plasma) and desirable and adverse effect curves, concentration may be substituted on the abscissa. Note that not all patients necessarily demonstrate a therapeutic response (or adverse effect) at any dose, and that some effects (notably some adverse effects) may occur in a dose-independent fashion.
PRINCIPLES OF PHARMACOKINETICS
The processes of absorption, distribution, metabolism, and excretion—collectively termed drug disposition—determine the concentration of drug delivered to target effector molecules.
ABSORPTION AND BIOAVAILABILITY
When a drug is administered orally, subcutaneously, intramuscularly, rectally, sublingually, or directly into desired sites of action, the amount of drug actually entering the systemic circulation may be less than with the intravenous route (Fig. 5-2A). The fraction of drug available to the systemic circulation by other routes is termed bioavailability. Bioavailability may be <100% for two main reasons: (1) absorption is reduced, or (2) the drug undergoes metabolism or elimination prior to entering the systemic circulation. Occasionally, the administered drug formulation is inconsistent or has degraded with time; for example, the anticoagulant dabigatran degrades rapidly (over weeks) once exposed to air, so the amount administered may be less than prescribed.
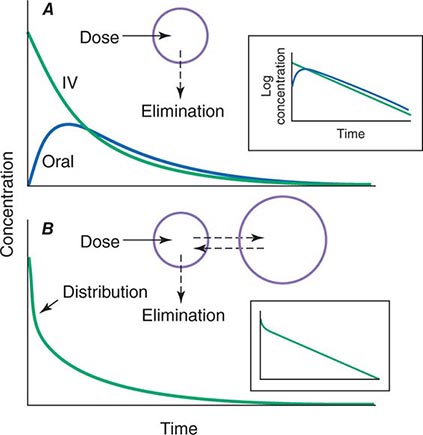
FIGURE 5-2 Idealized time-plasma concentration curves after a single dose of drug. A. The time course of drug concentration after an instantaneous IV bolus or an oral dose in the one-compartment model shown. The area under the time-concentration curve is clearly less with the oral drug than the IV, indicating incomplete bioavailability. Note that despite this incomplete bioavailability, concentration after the oral dose can be higher than after the IV dose at some time points. The inset shows that the decline of concentrations over time is linear on a log-linear plot, characteristic of first-order elimination, and that oral and IV drugs have the same elimination (parallel) time course. B. The decline of central compartment concentration when drug is distributed both to and from a peripheral compartment and eliminated from the central compartment. The rapid initial decline of concentration reflects not drug elimination but distribution.
When a drug is administered by a nonintravenous route, the peak concentration occurs later and is lower than after the same dose given by rapid intravenous injection, reflecting absorption from the site of administration (Fig. 5-2). The extent of absorption may be reduced because a drug is incompletely released from its dosage form, undergoes destruction at its site of administration, or has physicochemical properties such as insolubility that prevent complete absorption from its site of administration. Slow absorption rates are deliberately designed into “slow-release” or “sustained-release” drug formulations in order to minimize variation in plasma concentrations during the interval between doses.
“First-Pass” Effect When a drug is administered orally, it must traverse the intestinal epithelium, the portal venous system, and the liver prior to entering the systemic circulation (Fig. 5-3). Once a drug enters the enterocyte, it may undergo metabolism, be transported into the portal vein, or be excreted back into the intestinal lumen. Both excretion into the intestinal lumen and metabolism decrease systemic bioavailability. Once a drug passes this enterocyte barrier, it may also be taken up into the hepatocyte, where bioavailability can be further limited by metabolism or excretion into the bile. This elimination in intestine and liver, which reduces the amount of drug delivered to the systemic circulation, is termed presystemic elimination, presystemic extraction, or first-pass elimination.
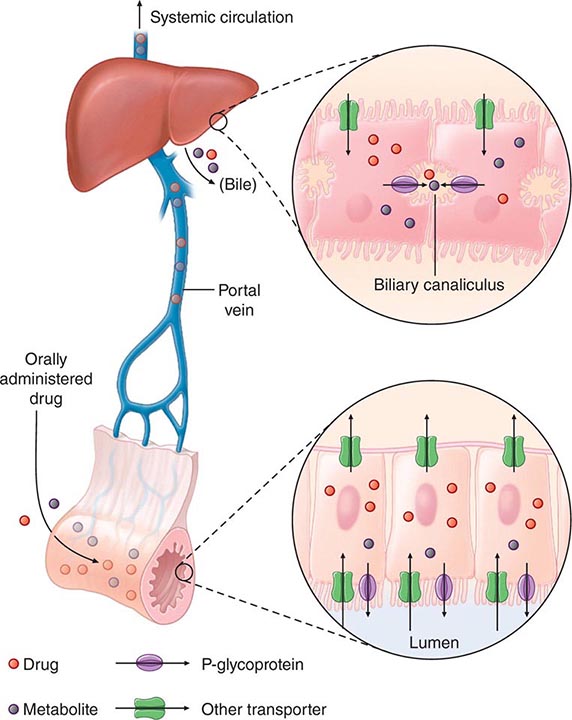
FIGURE 5-3 Mechanism of presystemic clearance. After drug enters the enterocyte, it can undergo metabolism, excretion into the intestinal lumen, or transport into the portal vein. Similarly, the hepatocyte may accomplish metabolism and biliary excretion prior to the entry of drug and metabolites to the systemic circulation. (Adapted by permission from DM Roden, in DP Zipes, J Jalife [eds]: Cardiac Electrophysiology: From Cell to Bedside, 4th ed. Philadelphia, Saunders, 2003. Copyright 2003 with permission from Elsevier.)
Drug movement across the membrane of any cell, including enterocytes and hepatocytes, is a combination of passive diffusion and active transport, mediated by specific drug uptake and efflux molecules. One widely studied drug transport molecule is P-glycoprotein, the product of the MDR1 gene. P-glycoprotein is expressed on the apical aspect of the enterocyte and on the canalicular aspect of the hepatocyte (Fig. 5-3). In both locations, it serves as an efflux pump, limiting availability of drug to the systemic circulation. P-glycoprotein–mediated drug efflux from cerebral capillaries limits drug brain penetration and is an important component of the blood-brain barrier.
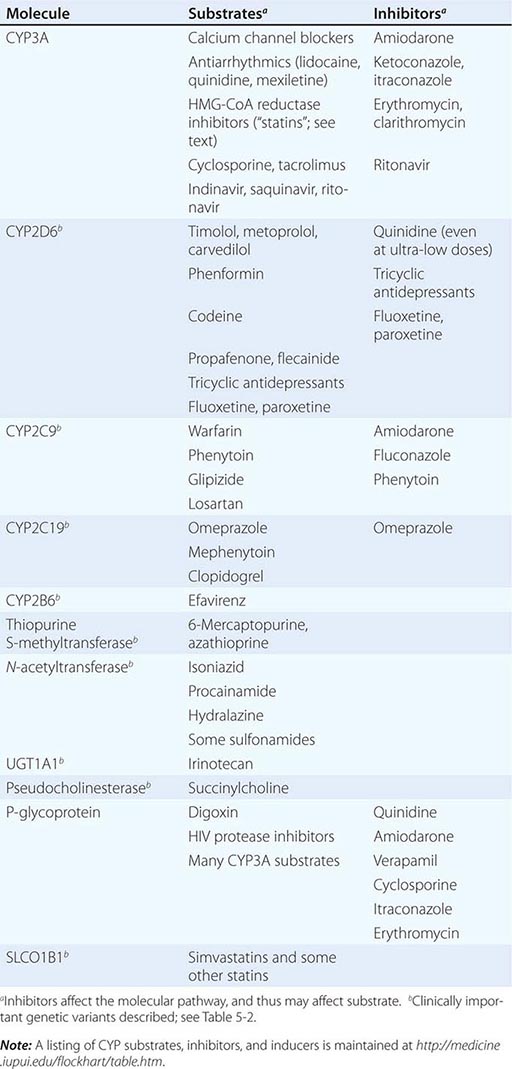
Drug metabolism generates compounds that are usually more polar and, hence, more readily excreted than parent drug. Metabolism takes place predominantly in the liver but can occur at other sites such as kidney, intestinal epithelium, lung, and plasma. “Phase I” metabolism involves chemical modification, most often oxidation accomplished by members of the cytochrome P450 (CYP) monooxygenase superfamily. CYPs that are especially important for drug metabolism are presented in Table 5-1, and each drug may be a substrate for one or more of these enzymes. “Phase II” metabolism involves conjugation of specific endogenous compounds to drugs or their metabolites. The enzymes that accomplish phase II reactions include glucuronyl-, acetyl-, sulfo-, and methyltransferases. Drug metabolites may exert important pharmacologic activity, as discussed further below.
MOLECULAR PATHWAYS MEDIATING DRUG DISPOSITION |
Clinical Implications of Altered Bioavailability Some drugs undergo near-complete presystemic metabolism and, thus, cannot be administered orally. Nitroglycerin cannot be used orally because it is completely extracted prior to reaching the systemic circulation. The drug is, therefore, used by the sublingual or transdermal routes, which bypass presystemic metabolism.
Some drugs with very extensive presystemic metabolism can still be administered by the oral route, using much higher doses than those required intravenously. Thus, a typical intravenous dose of verapamil is 1–5 mg, compared to the usual single oral dose of 40–120 mg. Administration of low-dose aspirin can result in exposure of cyclooxygenase in platelets in the portal vein to the drug, but systemic sparing because of first-pass aspirin deacylation in the liver. This is an example of presystemic metabolism being exploited to therapeutic advantage.
PHARMACOKINETIC CONCEPTS
Most pharmacokinetic processes, such as elimination, are first-order; that is, the rate of the process depends on the amount of drug present. Elimination can occasionally be zero-order (fixed amount eliminated per unit time), and this can be clinically important (see “Principles of Dose Selection”). In the simplest pharmacokinetic model (Fig. 5-2A), a drug bolus (D) is administered instantaneously to a central compartment, from which drug elimination occurs as a first-order process. Occasionally, central and other compartments correspond to physiologic spaces (e.g., plasma volume), whereas in others they are simply mathematical functions used to describe drug disposition. The first-order nature of drug elimination leads directly to the relationship describing drug concentration (C) at any time (t) following the bolus:
![]()
where VC is the volume of the compartment into which drug is delivered and t1/2 is elimination half-life. As a consequence of this relationship, a plot of the logarithm of concentration versus time is a straight line (Fig. 5-2A, inset). Half-life is the time required for 50% of a first-order process to be complete. Thus, 50% of drug elimination is achieved after one drug-elimination half-life, 75% after two, 87.5% after three, etc. In practice, first-order processes such as elimination are near-complete after four–five half-lives.
In some cases, drug is removed from the central compartment not only by elimination but also by distribution into peripheral compartments. In this case, the plot of plasma concentration versus time after a bolus may demonstrate two (or more) exponential components (Fig. 5-2B). In general, the initial rapid drop in drug concentration represents not elimination but drug distribution into and out of peripheral tissues (also first-order processes), while the slower component represents drug elimination; the initial precipitous decline is usually evident with administration by intravenous but not by other routes. Drug concentrations at peripheral sites are determined by a balance between drug distribution to and redistribution from those sites, as well as by elimination. Once distribution is near-complete (four–five distribution half-lives), plasma and tissue concentrations decline in parallel.
Clinical Implications of Half-Life Measurements The elimination half-life not only determines the time required for drug concentrations to fall to near-immeasurable levels after a single bolus, it is also the sole determinant of the time required for steady-state plasma concentrations to be achieved after any change in drug dosing (Fig. 5-4). This applies to the initiation of chronic drug therapy (whether by multiple oral doses or by continuous intravenous infusion), a change in chronic drug dose or dosing interval, or discontinuation of drug.
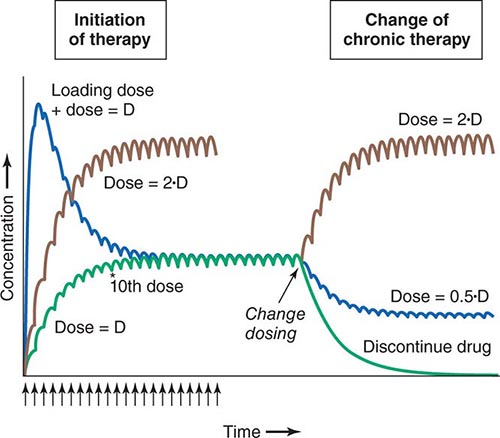
FIGURE 5-4 Drug accumulation to steady state. In this simulation, drug was administered (arrows) at intervals = 50% of the elimination half-life. Steady state is achieved during initiation of therapy after ~5 elimination half-lives, or 10 doses. A loading dose did not alter the eventual steady state achieved. A doubling of the dose resulted in a doubling of the steady state but the same time course of accumulation. Once steady state is achieved, a change in dose (increase, decrease, or drug discontinuation) results in a new steady state in ~5 elimination half-lives. (Adapted by permission from DM Roden, in DP Zipes, J Jalife [eds]: Cardiac Electrophysiology: From Cell to Bedside, 4th ed. Philadelphia, Saunders, 2003. Copyright 2003 with permission from Elsevier.)
Steady state describes the situation during chronic drug administration when the amount of drug administered per unit time equals drug eliminated per unit time. With a continuous intravenous infusion, plasma concentrations at steady state are stable, while with chronic oral drug administration, plasma concentrations vary during the dosing interval but the time-concentration profile between dosing intervals is stable (Fig. 5-4).
DRUG DISTRIBUTION
In a typical 70-kg human, plasma volume is ~3 L, blood volume is ~5.5 L, and extracellular water outside the vasculature is ~20 L. The volume of distribution of drugs extensively bound to plasma proteins but not to tissue components approaches plasma volume; warfarin is one such example. By contrast, for drugs highly bound to tissues, the volume of distribution can be far greater than any physiologic space. For example, the volume of distribution of digoxin and tricyclic antidepressants is hundreds of liters, obviously exceeding total-body volume. Such drugs are not readily removed by dialysis, an important consideration in overdose.
Clinical Implications of Drug Distribution In some cases, pharmacologic effects require drug distribution to peripheral sites. In this instance, the time course of drug delivery to and removal from these sites determines the time course of drug effects; anesthetic uptake into the central nervous system (CNS) is an example.
LOADING DOSES For some drugs, the indication may be so urgent that administration of “loading” dosages is required to achieve rapid elevations of drug concentration and therapeutic effects earlier than with chronic maintenance therapy (Fig. 5-4). Nevertheless, the time required for true steady state to be achieved is still determined only by the elimination half-life.
RATE OF INTRAVENOUS ADMINISTRATION Although the simulations in Fig. 5-2 use a single intravenous bolus, this is usually inappropriate in practice because side effects related to transiently very high concentrations can result. Rather, drugs are more usually administered orally or as a slower intravenous infusion. Some drugs are so predictably lethal when infused too rapidly that special precautions should be taken to prevent accidental boluses. For example, solutions of potassium for intravenous administration >20 mEq/L should be avoided in all but the most exceptional and carefully monitored circumstances. This minimizes the possibility of cardiac arrest due to accidental increases in infusion rates of more concentrated solutions.
Transiently high drug concentrations after rapid intravenous administration can occasionally be used to advantage. The use of midazolam for intravenous sedation, for example, depends upon its rapid uptake by the brain during the distribution phase to produce sedation quickly, with subsequent egress from the brain during the redistribution of the drug as equilibrium is achieved.
Similarly, adenosine must be administered as a rapid bolus in the treatment of reentrant supraventricular tachycardias (Chap. 276) to prevent elimination by very rapid (t1/2 of seconds) uptake into erythrocytes and endothelial cells before the drug can reach its clinical site of action, the atrioventricular node.
Clinical Implications of Altered Protein Binding Many drugs circulate in the plasma partly bound to plasma proteins. Since only unbound (free) drug can distribute to sites of pharmacologic action, drug response is related to the free rather than the total circulating plasma drug concentration. In chronic kidney or liver disease, protein binding may be decreased and thus drug actions increased. In some situations (myocardial infarction, infection, surgery), acute phase reactants transiently increase drug binding and thus decrease efficacy. These changes assume the greatest clinical importance for drugs that are highly protein-bound since even a small change in protein binding can result in large changes in free drug; for example, a decrease in binding from 99% to 98% doubles the free drug concentration from 1% to 2%. For some drugs (e.g., phenytoin), monitoring free rather than total drug concentrations can be useful.
ELIMINATION
Drug elimination reduces the amount of drug in the body over time. An important approach to quantifying this reduction is to consider that drug concentrations at the beginning and end of a time period are unchanged and that a specific volume of the body has been “cleared” of the drug during that time period. This defines clearance as volume/time. Clearance includes both drug metabolism and excretion.
Clinical Implications of Altered Clearance While elimination half-life determines the time required to achieve steady-state plasma concentration (Css), the magnitude of that steady state is determined by clearance (Cl) and dose alone. For a drug administered as an intravenous infusion, this relationship is:
Css = dosing rate/Cl or dosing rate = Cl · Css
When drug is administered orally, the average plasma concentration within a dosing interval (Cavg,ss) replaces Css, and the dosage (dose per unit time) must be increased if bioavailability (F) is less than 1:
Dose/time = Cl · Cavg,ss/F
Genetic variants, drug interactions, or diseases that reduce the activity of drug-metabolizing enzymes or excretory mechanisms lead to decreased clearance and, hence, a requirement for downward dose adjustment to avoid toxicity. Conversely, some drug interactions and genetic variants increase the function of drug elimination pathways, and hence, increased drug dosage is necessary to maintain a therapeutic effect.
ACTIVE DRUG METABOLITES
Metabolites may produce effects similar to, overlapping with, or distinct from those of the parent drug. Accumulation of the major metabolite of procainamide, N-acetylprocainamide (NAPA), likely accounts for marked QT prolongation and torsades des pointes ventricular tachycardia (Chap. 276) during therapy with procainamide. Neurotoxicity during therapy with the opioid analgesic meperidine is likely due to accumulation of normeperidine, especially in renal disease.
Prodrugs are inactive compounds that require metabolism to generate active metabolites that mediate the drug effects. Examples include many angiotensin-converting enzyme (ACE) inhibitors, the angiotensin receptor blocker losartan, the antineoplastic irinotecan, the anti-estrogen tamoxifen, the analgesic codeine (whose active metabolite morphine probably underlies the opioid effect during codeine administration), and the antiplatelet drug clopidogrel. Drug metabolism has also been implicated in bioactivation of procarcinogens and in generation of reactive metabolites that mediate certain adverse drug effects (e.g., acetaminophen hepatotoxicity, discussed below).
THE CONCEPT OF HIGH-RISK PHARMACOKINETICS
When plasma concentrations of active drug depend exclusively on a single metabolic pathway, any condition that inhibits that pathway (be it disease-related, genetic, or due to a drug interaction) can lead to dramatic changes in drug concentrations and marked variability in drug action. This problem of high-risk pharmacokinetics is especially pronounced in two settings. First, variability in bioactivation of a prodrug can lead to striking variability in drug action; examples include decreased CYP2D6 activity, which prevents analgesia by codeine, and decreased CYP2C19 activity, which reduces the antiplatelet effects of clopidogrel. The second setting is drug elimination that relies on a single pathway. In this case, inhibition of the elimination pathway by genetic variants or by administration of inhibiting drugs leads to marked elevation of drug concentration and, for drugs with a narrow therapeutic window, an increased likelihood of dose-related toxicity. Individuals with loss-of-function alleles in CYP2C9, responsible for metabolism of the active S-enantiomer of warfarin, appear to be at increased risk for bleeding. When drugs undergo elimination by multiple-drug metabolizing or excretory pathways, absence of one pathway (due to a genetic variant or drug interaction) is much less likely to have a large impact on drug concentrations or drug actions.
PRINCIPLES OF PHARMACODYNAMICS
The Onset of Drug Action For drugs used in the urgent treatment of acute symptoms, little or no delay is anticipated (or desired) between the drug-target interaction and the development of a clinical effect. Examples of such acute situations include vascular thrombosis, shock, or status epilepticus.
For many conditions, however, the indication for therapy is less urgent, and a delay between the interaction of a drug with its pharmacologic target(s) and a clinical effect is clinically acceptable. Common pharmacokinetic mechanisms that can contribute to such a delay include slow elimination (resulting in slow accumulation to steady state), uptake into peripheral compartments, or accumulation of active metabolites. Another common explanation for such a delay is that the clinical effect develops as a downstream consequence of the initial molecular effect the drug produces. Thus, administration of a proton pump inhibitor or an H2-receptor blocker produces an immediate increase in gastric pH but ulcer healing that is delayed. Cancer chemotherapy similarly produces delayed therapeutic effects.
Drug Effects May Be Disease Specific A drug may produce no action or a different spectrum of actions in unaffected individuals compared to patients with underlying disease. Further, concomitant disease can complicate interpretation of response to drug therapy, especially adverse effects. For example, high doses of anticonvulsants such as phenytoin may cause neurologic symptoms, which may be confused with the underlying neurologic disease. Similarly, increasing dyspnea in a patient with chronic lung disease receiving amiodarone therapy could be due to drug, underlying disease, or an intercurrent cardiopulmonary problem. Thus, the presence of chronic lung disease may argue against the use of amiodarone.
While drugs interact with specific molecular receptors, drug effects may vary over time, even if stable drug and metabolite concentrations are maintained. The drug-receptor interaction occurs in a complex biologic milieu that can vary to modulate the drug effect. For example, ion channel blockade by drugs, an important anticonvulsant and antiarrhythmic effect, is often modulated by membrane potential, itself a function of factors such as extracellular potassium or local ischemia. Receptors may be up- or downregulated by disease or by the drug itself. For example, β-adrenergic blockers upregulate β-receptor density during chronic therapy. While this effect does not usually result in resistance to the therapeutic effect of the drugs, it may produce severe agonist-mediated effects (such as hypertension or tachycardia) if the blocking drug is abruptly withdrawn.
PRINCIPLES OF DOSE SELECTION
The desired goal of therapy with any drug is to maximize the likelihood of a beneficial effect while minimizing the risk of adverse effects. Previous experience with the drug, in controlled clinical trials or in postmarketing use, defines the relationships between dose or plasma concentration and these dual effects (Fig. 5-1) and has important implications for initiation of drug therapy:
1. The target drug effect should be defined when drug treatment is started. With some drugs, the desired effect may be difficult to measure objectively, or the onset of efficacy can be delayed for weeks or months; drugs used in the treatment of cancer and psychiatric disease are examples. Sometimes a drug is used to treat a symptom, such as pain or palpitations, and here it is the patient who will report whether the selected dose is effective. In yet other settings, such as anticoagulation or hypertension, the desired response can be repeatedly and objectively assessed by simple clinical or laboratory tests.
2. The nature of anticipated toxicity often dictates the starting dose. If side effects are minor, it may be acceptable to start chronic therapy at a dose highly likely to achieve efficacy and down-titrate if side effects occur. However, this approach is rarely, if ever, justified if the anticipated toxicity is serious or life-threatening; in this circumstance, it is more appropriate to initiate therapy with the lowest dose that may produce a desired effect. In cancer chemotherapy, it is common practice to use maximum-tolerated doses.
3. The above considerations do not apply if these relationships between dose and effects cannot be defined. This is especially relevant to some adverse drug effects (discussed in further detail below) whose development are not readily related to drug dose.
4. If a drug dose does not achieve its desired effect, a dosage increase is justified only if toxicity is absent and the likelihood of serious toxicity is small.
Failure of Efficacy Assuming the diagnosis is correct and the correct drug is prescribed, explanations for failure of efficacy include drug interactions, noncompliance, or unexpectedly low drug dosage due to administration of expired or degraded drug. These are situations in which measurement of plasma drug concentrations, if available, can be especially useful. Noncompliance is an especially frequent problem in the long-term treatment of diseases such as hypertension and epilepsy, occurring in ≥25% of patients in therapeutic environments in which no special effort is made to involve patients in the responsibility for their own health. Multidrug regimens with multiple doses per day are especially prone to noncompliance.
Monitoring response to therapy, by physiologic measures or by plasma concentration measurements, requires an understanding of the relationships between plasma concentration and anticipated effects. For example, measurement of QT interval is used during treatment with sotalol or dofetilide to avoid marked QT prolongation that can herald serious arrhythmias. In this setting, evaluating the electrocardiogram at the time of anticipated peak plasma concentration and effect (e.g., 1–2 h postdose at steady state) is most appropriate. Maintained high vancomycin levels carry a risk of nephrotoxicity, so dosages should be adjusted on the basis of plasma concentrations measured at trough (predose). Similarly, for dose adjustment of other drugs (e.g., anticonvulsants), concentration should be measured at its lowest during the dosing interval, just prior to a dose at steady state (Fig. 5-4), to ensure a maintained therapeutic effect.
Concentration of Drugs in Plasma as a Guide to Therapy Factors such as interactions with other drugs, disease-induced alterations in elimination and distribution, and genetic variation in drug disposition combine to yield a wide range of plasma levels in patients given the same dose. Hence, if a predictable relationship can be established between plasma drug concentration and beneficial or adverse drug effect, measurement of plasma levels can provide a valuable tool to guide selection of an optimal dose, especially when there is a narrow range between the plasma levels yielding therapeutic and adverse effects. Monitoring is commonly used with certain types of drugs including many anticonvulsants, antirejection agents, antiarrhythmics, and antibiotics. By contrast, if no such relationship can be established (e.g., if drug access to important sites of action outside plasma is highly variable), monitoring plasma concentration may not provide an accurate guide to therapy (Fig. 5-5A).
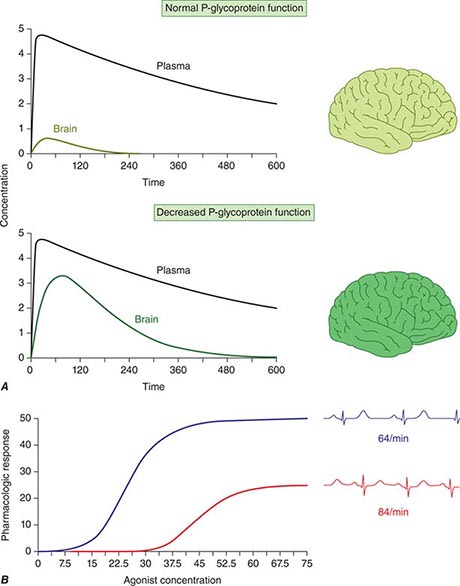
FIGURE 5-5 A. The efflux pump P-glycoprotein excludes drugs from the endothelium of capillaries in the brain and so constitutes a key element of the blood-brain barrier. Thus, reduced P-glycoprotein function (e.g., due to drug interactions or genetically determined variability in gene transcription) increases penetration of substrate drugs into the brain, even when plasma concentrations are unchanged. B. The graph shows an effect of a β1-receptor polymorphism on receptor function in vitro. Patients with the hypofunctional variant (red) may display lesser heart-rate slowing or blood pressure lowering on exposure to a receptor blocking agent.
The common situation of first-order elimination implies that average, maximum, and minimum steady-state concentrations are related linearly to the dosing rate. Accordingly, the maintenance dose may be adjusted on the basis of the ratio between the desired and measured concentrations at steady state; for example, if a doubling of the steady-state plasma concentration is desired, the dose should be doubled. This does not apply to drugs eliminated by zero-order kinetics (fixed amount per unit time), where small dosage increases will produce disproportionate increases in plasma concentration; examples include phenytoin and theophylline.
An increase in dosage is usually best achieved by changing the drug dose but not the dosing interval (e.g., by giving 200 mg every 8 h instead of 100 mg every 8 h). However, this approach is acceptable only if the resulting maximum concentration is not toxic and the trough value does not fall below the minimum effective concentration for an undesirable period of time. Alternatively, the steady state may be changed by altering the frequency of intermittent dosing but not the size of each dose. In this case, the magnitude of the fluctuations around the average steady-state level will change—the shorter the dosing interval, the smaller the difference between peak and trough levels.
EFFECTS OF DISEASE ON DRUG CONCENTRATION AND RESPONSE
RENAL DISEASE
Renal excretion of parent drug and metabolites is generally accomplished by glomerular filtration and by specific drug transporters. If a drug or its metabolites are primarily excreted through the kidneys and increased drug levels are associated with adverse effects, drug dosages must be reduced in patients with renal dysfunction to avoid toxicity. The antiarrhythmics dofetilide and sotalol undergo predominant renal excretion and carry a risk of QT prolongation and arrhythmias if doses are not reduced in renal disease. In end-stage renal disease, sotalol has been given as 40 mg after dialysis (every second day), compared to the usual daily dose, 80–120 mg every 12 h. The narcotic analgesic meperidine undergoes extensive hepatic metabolism, so that renal failure has little effect on its plasma concentration. However, its metabolite, normeperidine, does undergo renal excretion, accumulates in renal failure, and probably accounts for the signs of CNS excitation, such as irritability, twitching, and seizures, that appear when multiple doses of meperidine are administered to patients with renal disease. Protein binding of some drugs (e.g., phenytoin) may be altered in uremia, so measuring free drug concentration may be desirable.
In non-end-stage renal disease, changes in renal drug clearance are generally proportional to those in creatinine clearance, which may be measured directly or estimated from the serum creatinine (Chap. 333e). This estimate, coupled with the knowledge of how much drug is normally excreted renally versus nonrenally, allows an estimate of the dose adjustment required. In practice, most decisions involving dosing adjustment in patients with renal failure use published recommended adjustments in dosage or dosing interval based on the severity of renal dysfunction indicated by creatinine clearance. Any such modification of dose is a first approximation and should be followed by plasma concentration data (if available) and clinical observation to further optimize therapy for the individual patient.
LIVER DISEASE
Standard tests of liver function are not useful in adjusting doses in diseases like hepatitis or cirrhosis. First-pass metabolism may decrease, leading to increased oral bioavailability as a consequence of disrupted hepatocyte function, altered liver architecture, and portacaval shunts. The oral bioavailability for high first-pass drugs such as morphine, meperidine, midazolam, and nifedipine is almost doubled in patients with cirrhosis, compared to those with normal liver function. Therefore, the size of the oral dose of such drugs should be reduced in this setting.
HEART FAILURE AND SHOCK
Under conditions of decreased tissue perfusion, the cardiac output is redistributed to preserve blood flow to the heart and brain at the expense of other tissues (Chap. 279). As a result, drugs may be distributed into a smaller volume of distribution, higher drug concentrations will be present in the plasma, and the tissues that are best perfused (the brain and heart) will be exposed to these higher concentrations, resulting in increased CNS or cardiac effects. As well, decreased perfusion of the kidney and liver may impair drug clearance. Another consequence of severe heart failure is decreased gut perfusion, which may reduce drug absorption and, thus, lead to reduced or absent effects of orally administered therapies.
DRUG USE IN THE ELDERLY
In the elderly, multiple pathologies and medications used to treat them result in more drug interactions and adverse effects. Aging also results in changes in organ function, especially of the organs involved in drug disposition. Initial doses should be less than the usual adult dosage and should be increased slowly. The number of medications, and doses per day, should be kept as low as possible.
Even in the absence of kidney disease, renal clearance may be reduced by 35–50% in elderly patients. Dosages should be adjusted on the basis of creatinine clearance. Aging also results in a decrease in the size of, and blood flow to, the liver and possibly in the activity of hepatic drug-metabolizing enzymes; accordingly, the hepatic clearance of some drugs is impaired in the elderly. As with liver disease, these changes are not readily predicted.
Elderly patients may display altered drug sensitivity. Examples include increased analgesic effects of opioids, increased sedation from benzodiazepines and other CNS depressants, and increased risk of bleeding while receiving anticoagulant therapy, even when clotting parameters are well controlled. Exaggerated responses to cardiovascular drugs are also common because of the impaired responsiveness of normal homeostatic mechanisms. Conversely, the elderly display decreased sensitivity to β-adrenergic receptor blockers.
Adverse drug reactions are especially common in the elderly because of altered pharmacokinetics and pharmacodynamics, the frequent use of multidrug regimens, and concomitant disease. For example, use of long half-life benzodiazepines is linked to the occurrence of hip fractures in elderly patients, perhaps reflecting both a risk of falls from these drugs (due to increased sedation) and the increased incidence of osteoporosis in elderly patients. In population surveys of the noninstitutionalized elderly, as many as 10% had at least one adverse drug reaction in the previous year.
DRUG USE IN CHILDREN
While most drugs used to treat disease in children are the same are those in adults, there are few studies that provide solid data to guide dosing. Drug metabolism pathways mature at different rates after birth, and disease mechanisms may be different in children. In practice, doses are adjusted for size (weight or body surface area) as a first approximation unless age-specific data are available.
GENETIC DETERMINANTS OF THE RESPONSE TO DRUGS
PRINCIPLES OF GENETIC VARIATION AND HUMAN TRAITS (See also CHAPS. 82 AND 84)
The concept that genetically determined variations in drug metabolism might be associated with variable drug levels and hence, effect, was advanced at the end of the nineteenth century, and the examples of familial clustering of unusual drug responses were noted in the mid-twentieth century. A goal of traditional Mendelian genetics is to identify DNA variants associated with a distinct phenotype in multiple related family members (Chap. 84). However, it is unusual for a drug response phenotype to be accurately measured in more than one family member, let alone across a kindred. Thus, non-family-based approaches are generally used to identify and validate DNA variants contributing to variable drug actions.
Candidate Gene Studies in Pharmacogenetics Most studies to date have used an understanding of the molecular mechanisms modulating drug action to identify candidate genes in which variants could explain variable drug responses. One very common scenario is that variable drug actions can be attributed to variability in plasma drug concentrations. When plasma drug concentrations vary widely (e.g., more than an order of magnitude), especially if their distribution is non-unimodal as in Fig. 5-6, variants in single genes controlling drug concentrations often contribute. In this case, the most obvious candidate genes are those responsible for drug metabolism and elimination. Other candidate genes are those encoding the target molecules with which drugs interact to produce their effects or molecules modulating that response, including those involved in disease pathogenesis.
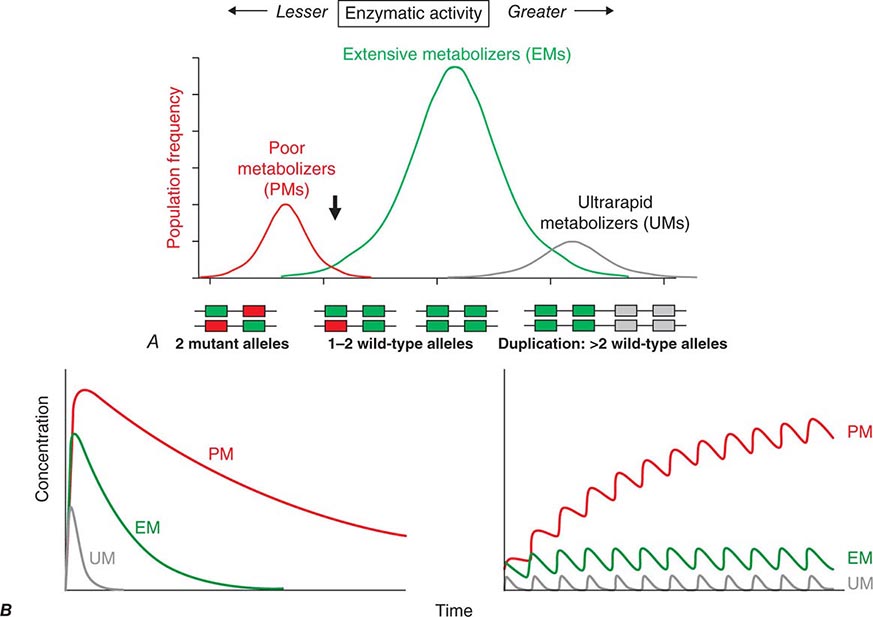
FIGURE 5-6 A. CYP2D6 metabolic activity was assessed in 290 subjects by administration of a test dose of a probe substrate and measurement of urinary formation of the CYP2D6-generated metabolite. The heavy arrow indicates a clear antimode, separating poor metabolizer subjects (PMs, red), with two loss-of-function CYP2D6 alleles, indicated by the intron-exon structures below the chart. Individuals with one or two functional alleles are grouped together as extensive metabolizers (EMs, green). Also shown are ultra-rapid metabolizers (UMs), with 2–12 functional copies of the gene (gray), displaying the greatest enzyme activity. (Adapted from M-L Dahl et al: J Pharmacol Exp Ther 274:516, 1995.) B. These simulations show the predicted effects of CYP2D6 genotype on disposition of a substrate drug. With a single dose (left), there is an inverse “gene-dose” relationship between the number of active alleles and the areas under the time-concentration curves (smallest in UM subjects; highest in PM subjects); this indicates that clearance is greatest in UM subjects. In addition, elimination half-life is longest in PM subjects. The right panel shows that these single dose differences are exaggerated during chronic therapy: steady-state concentration is much higher in PM subjects (decreased clearance), as is the time required to achieve steady state (longer elimination half-life).
Genome-Wide Association Studies in Pharmacogenomics The field has also had some success with “unbiased” approaches such as genome-wide association (GWA) (Chap. 82), particularly in identifying single variants associated with high risk for certain forms of drug toxicity (Table 5-2). GWA studies have identified variants in the HLA-B locus that are associated with high risk for severe skin rashes during treatment with the anticonvulsant carbamazepine and the antiretroviral abacavir. A GWA study of simvastatin-associated myopathy identified a single noncoding single nucleotide polymorphism (SNP) in SLCO1B1, encoding OATP1B1, a drug transporter known to modulate simvastatin uptake into the liver, which accounts for 60% of myopathy risk. GWA approaches have also implicated interferon variants in antileukemic responses and in response to therapy in hepatitis C. Ribavirin, used as therapy in hepatitis C, causes hemolytic anemia, and this has been linked to variants in ITPA, encoding inosine triphosphatase.
GENETIC VARIANTS AND DRUG RESPONSES |
GENETIC VARIANTS AFFECTING PHARMACOKINETICS
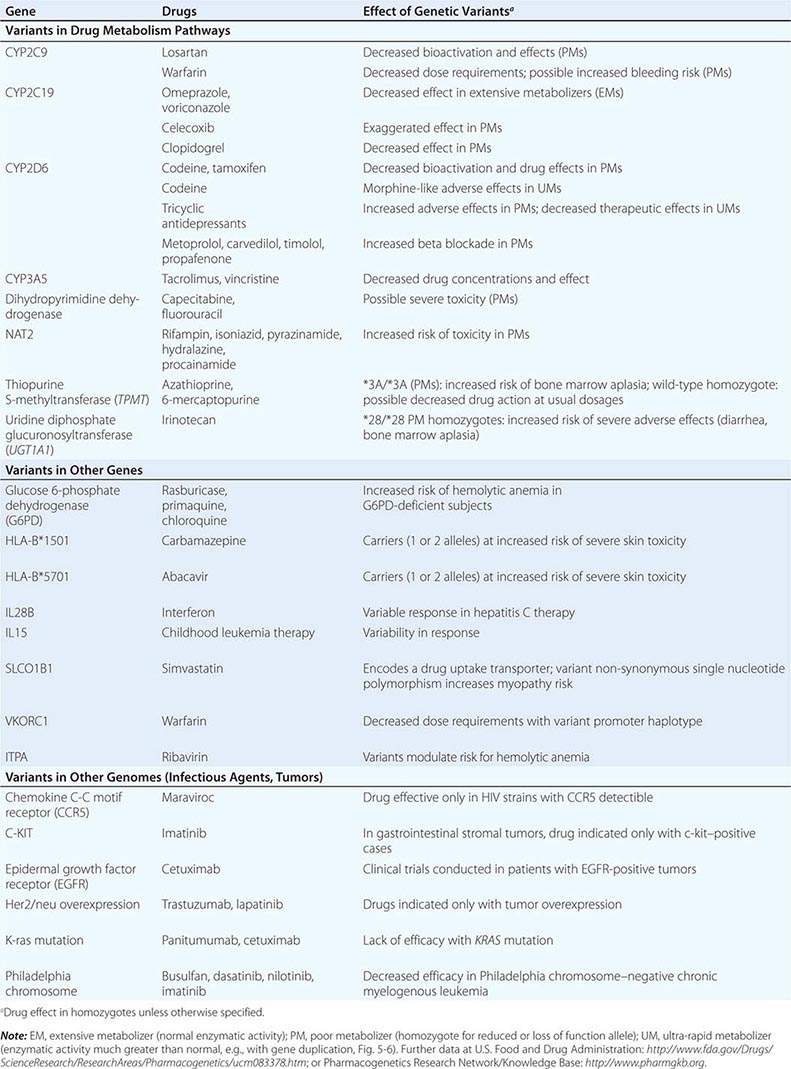
Clinically important genetic variants have been described in multiple molecular pathways of drug disposition (Table 5-2). A distinct multimodal distribution of drug disposition (as shown in Fig. 5-6) argues for a predominant effect of variants in a single gene in the metabolism of that substrate. Individuals with two alleles (variants) encoding for nonfunctional protein make up one group, often termed poor metabolizers (PM phenotype); for some genes, many variants can produce such a loss of function, complicating the use of genotyping in clinical practice. Individuals with one functional allele make up a second (intermediate metabolizers) and may or may not be distinguishable from those with two functional alleles (extensive metabolizers, EMs). Ultra-rapid metabolizers with especially high enzymatic activity (occasionally due to gene duplication; Fig. 5-6) have also been described for some traits. Many drugs in widespread use can inhibit specific drug disposition pathways (Table 5-1), and so EM individuals receiving such inhibitors can respond like PM patients (phenocopying). Polymorphisms in genes encoding drug uptake or drug efflux transporters may be other contributors to variability in drug delivery to target sites and, hence, in drug effects.
CYP Variants Members of the CYP3A family (CYP3A4, 3A5) metabolize the greatest number of drugs in therapeutic use. CYP3A4 activity is highly variable (up to an order of magnitude) among individuals, but the underlying mechanisms are not well understood. In whites, but not African Americans, there is a common loss-of-function polymorphism in the closely related CYP3A5 gene. Decreased efficacy of the antirejection agent tacrolimus in African-American subjects has been attributed to more rapid elimination due to relatively greater CYP3A5 activity. A lower risk of vincristine-associated neuropathy has been reported in CYP3A5 “expressers.”
CYP2D6 is second to CYP3A4 in the number of commonly used drugs that it metabolizes. CYP2D6 activity is polymorphically distributed, with about 7% of European- and African-derived populations (but very few Asians) displaying the PM phenotype (Fig. 5-6). Dozens of loss-of-function variants in the CYP2D6 gene have been described; the PM phenotype arises in individuals with two such alleles. In addition, ultra-rapid metabolizers with multiple functional copies of the CYP2D6 gene have been identified.
Codeine is biotransformed by CYP2D6 to the potent active metabolite morphine, so its effects are blunted in PMs and exaggerated in ultra-rapid metabolizers. In the case of drugs with beta-blocking properties metabolized by CYP2D6, greater signs of beta blockade (e.g., bronchospasm, bradycardia) are seen in PM subjects than in EMs. This can be seen not only with orally administered beta blockers such as metoprolol and carvedilol, but also with ophthalmic timolol and with the sodium channel–blocking antiarrhythmic propafenone, a CYP2D6 substrate with beta-blocking properties. Ultra-rapid metabolizers may require very high dosages of tricyclic antidepressants to achieve a therapeutic effect and, with codeine, may display transient euphoria and nausea due to very rapid generation of morphine. Tamoxifen undergoes CYP2D6-mediated biotransformation to an active metabolite, so its efficacy may be in part related to this polymorphism. In addition, the widespread use of selective serotonin reuptake inhibitors (SSRIs) to treat tamoxifen-related hot flashes may also alter the drug’s effects because many SSRIs, notably fluoxetine and paroxetine, are also CYP2D6 inhibitors.
The PM phenotype for CYP2C19 is common (20%) among Asians and rarer (2–3%) in European-derived populations. The impact of polymorphic CYP2C19-mediated metabolism has been demonstrated with the proton pump inhibitor omeprazole, where ulcer cure rates with “standard” dosages were much lower in EM patients (29%) than in PMs (100%). Thus, understanding the importance of this polymorphism would have been important in developing the drug, and knowing a patient’s CYP2C19 genotype should improve therapy. CYP2C19 is responsible for bioactivation of the antiplatelet drug clopidogrel, and several large studies have documented decreased efficacy (e.g., increased myocardial infarction after placement of coronary stents) among Caucasian subjects with reduction of function alleles. In addition, some studies suggest that omeprazole and possibly other proton pump inhibitors phenocopy this effect.
There are common variants of CYP2C9 that encode proteins with loss of catalytic function. These variant alleles are associated with increased rates of neurologic complications with phenytoin, hypoglycemia with glipizide, and reduced warfarin dose required to maintain stable anticoagulation. The angiotensin-receptor blocker losartan is a prodrug that is bioactivated by CYP2C9; as a result, PMs and those receiving inhibitor drugs may display little response to therapy.
Transferase Variants One of the most extensively studied phase II polymorphisms is the PM trait for thiopurine S-methyltransferase (TPMT). TPMT bioinactivates the antileukemic drug 6-mercaptopurine. Further, 6-mercaptopurine is itself an active metabolite of the immunosuppressive azathioprine. Homozygotes for alleles encoding the inactive TPMT (1 in 300 individuals) predictably exhibit severe and potentially fatal pancytopenia on standard doses of azathioprine or 6-mercaptopurine. On the other hand, homozygotes for fully functional alleles may display less anti-inflammatory or antileukemic effect with the drugs.
N-acetylation is catalyzed by hepatic N-acetyl transferase (NAT), which represents the activity of two genes, NAT-1 and NAT-2. Both enzymes transfer an acetyl group from acetyl coenzyme A to the drug; polymorphisms in NAT-2 are thought to underlie individual differences in the rate at which drugs are acetylated and thus define “rapid acetylators” and “slow acetylators.” Slow acetylators make up ~50% of European- and African-derived populations but are less common among Asians. Slow acetylators have an increased incidence of the drug-induced lupus syndrome during procainamide and hydralazine therapy and of hepatitis with isoniazid. Induction of CYPs (e.g., by rifampin) also increases the risk of isoniazid-related hepatitis, likely reflecting generation of reactive metabolites of acetylhydrazine, itself an isoniazid metabolite.
Individuals homozygous for a common promoter polymorphism that reduces transcription of uridine diphosphate glucuronosyltransferase (UGT1A1) have benign hyperbilirubinemia (Gilbert’s syndrome; Chap. 358). This variant has also been associated with diarrhea and increased bone marrow depression with the antineoplastic prodrug irinotecan, whose active metabolite is normally detoxified by UGT1A1-mediated glucuronidation. The antiretroviral atazanavir is a UGT1A1 inhibitor, and individuals with the Gilbert’s variant develop higher bilirubin levels during treatment.
VARIABILITY IN THE MOLECULAR TARGETS WITH WHICH DRUGS INTERACT
Multiple polymorphisms identified in the β2-adrenergic receptor appear to be linked to specific phenotypes in asthma and congestive heart failure, diseases in which β2-receptor function might be expected to determine prognosis. Polymorphisms in the β2-receptor gene have also been associated with response to inhaled β2-receptor agonists, while those in the β1-adrenergic receptor gene have been associated with variability in heart rate slowing and blood pressure lowering (Fig. 5-5B). In addition, in heart failure, a common polymorphism in the β1-adrenergic receptor gene has been implicated in variable clinical outcome during therapy with the investigational beta blocker bucindolol. Response to the 5-lipoxygenase inhibitor zileuton in asthma has been linked to polymorphisms that determine the expression level of the 5-lipoxygenase gene.
Drugs may also interact with genetic pathways of disease to elicit or exacerbate symptoms of the underlying conditions. In the porphyrias, CYP inducers are thought to increase the activity of enzymes proximal to the deficient enzyme, exacerbating or triggering attacks (Chap. 430). Deficiency of glucose-6-phosphate dehydrogenase (G6PD), most often in individuals of African, Mediterranean, or South Asian descent, increases the risk of hemolytic anemia in response to the antimalarial primaquine (Chap. 129) and the uric acid–lowering agent rasburicase, which do not cause hemolysis in patients with normal amounts of the enzyme. Patients with mutations in the ryanodine receptor, which controls intracellular calcium in skeletal muscle and other tissues, may be asymptomatic until exposed to certain general anesthetics, which trigger the rare syndrome of malignant hyperthermia. Certain antiarrhythmics and other drugs can produce marked QT prolongation and torsades des pointes (Chap. 276), and in some patients, this adverse effect represents unmasking of previously subclinical congenital long QT syndrome. Up to 50% of the variability in steady-state warfarin dose requirement is attributable to polymorphisms in the promoter of VKORC1, which encodes the warfarin target, and in the coding region of CYP2C9, which mediates its elimination.
Tumor and Infectious Agent Genomes The actions of drugs used to treat infectious or neoplastic disease may be modulated by variants in these nonhuman germline genomes. Genotyping tumors is a rapidly evolving approach to target therapies to underlying mechanisms and to avoid potentially toxic therapy in patients who would derive no benefit (Chap. 101e). Trastuzumab, which potentiates anthracycline-related cardiotoxicity, is ineffective in breast cancers that do not express the herceptin receptor. Imatinib targets a specific tyrosine kinase, BCR-Abl1, that is generated by the translocation that creates the Philadelphia chromosome typical of chronic myelogenous leukemia (CML). BCR-Abl1 is not only active but may be central to the pathogenesis of CML; its use in BCR-Abl1-positive tumors has resulted in remarkable antitumor efficacy. Similarly, the anti–epidermal growth factor receptor (EGFR) antibodies cetuximab and panitumumab appear especially effective in colon cancers in which K-ras, a G protein in the EGFR pathway, is not mutated. Vemurafenib does not inhibit wild-type BRAF but is active against the V600E mutant form of the kinase.
PROSPECTS FOR INCORPORATING PHARMACOGENETIC INFORMATION INTO CLINICAL PRACTICE
The description of genetic variants linked to variable drug responses naturally raises the question of if and how to use this information in practice. Indeed, the U.S. Food and Drug Administration (FDA) now incorporates pharmacogenetic data into information (“package inserts”) meant to guide prescribing. A decision to adopt pharmacogenetically guided dosing for a given drug depends on multiple factors. The most important are the magnitude and clinical importance of the genetic effect and the strength of evidence linking genetic variation to variable drug effects (e.g., anecdote versus post-hoc analysis of clinical trial data versus randomized prospective clinical trial). The evidence can be strengthened if statistical arguments from clinical trial data are complemented by an understanding of underlying physiologic mechanisms. Cost versus expected benefit may also be a factor.
When the evidence is compelling, alternate therapies are not available, and there are clear recommendations for dosage adjustment in subjects with variants, there is a strong argument for deploying genetic testing as a guide to prescribing. The association between HLA-B*5701 and severe skin toxicity with abacavir is an example. In other situations, the arguments are less compelling: the magnitude of the genetic effect may be smaller, the consequences may be less serious, alternate therapies may be available, or the drug effect may be amenable to monitoring by other approaches. Ongoing clinical trials are addressing the utility of preprescription genotyping in large populations exposed to drugs with known pharmacogenetic variants (e.g., warfarin). Importantly, technological advances are now raising the possibility of inexpensive whole genome sequencing. Incorporating a patient’s whole genome sequence into their electronic medical record would allow the information to be accessed as needed for many genetic and pharmacogenetic applications, and the argument has been put forward that this approach would lower logistic barriers to use of pharmacogenomic variant data in prescribing. There are multiple issues (e.g., economic, technological, and ethical) that need to be addressed if such a paradigm is to be adopted (Chap. 82). While barriers to bringing genomic and pharmacogenomic information to the bedside seem daunting, the field is very young and evolving rapidly. Indeed, one major result of understanding the role of genetics in drug action has been improved screening of drugs during the development process to reduce the likelihood of highly variable metabolism or unanticipated toxicity.
INTERACTIONS BETWEEN DRUGS
Drug interactions can complicate therapy by increasing or decreasing the action of a drug; interactions may be based on changes in drug disposition or in drug response in the absence of changes in drug levels. Interactions must be considered in the differential diagnosis of any unusual response occurring during drug therapy. Prescribers should recognize that patients often come to them with a legacy of drugs acquired during previous medical experiences, often with multiple physicians who may not be aware of all the patient’s medications. A meticulous drug history should include examination of the patient’s medications and, if necessary, calls to the pharmacist to identify prescriptions. It should also address the use of agents not often volunteered during questioning, such as OTC drugs, health food supplements, and topical agents such as eye drops. Lists of interactions are available from a number of electronic sources. While it is unrealistic to expect the practicing physician to memorize these, certain drugs consistently run the risk of generating interactions, often by inhibiting or inducing specific drug elimination pathways. Examples are presented below and in Table 5-3. Accordingly, when these drugs are started or stopped, prescribers must be especially alert to the possibility of interactions.
DRUGS WITH A HIGH RISK OF GENERATING PHARMACOKINETIC INTERACTIONS |
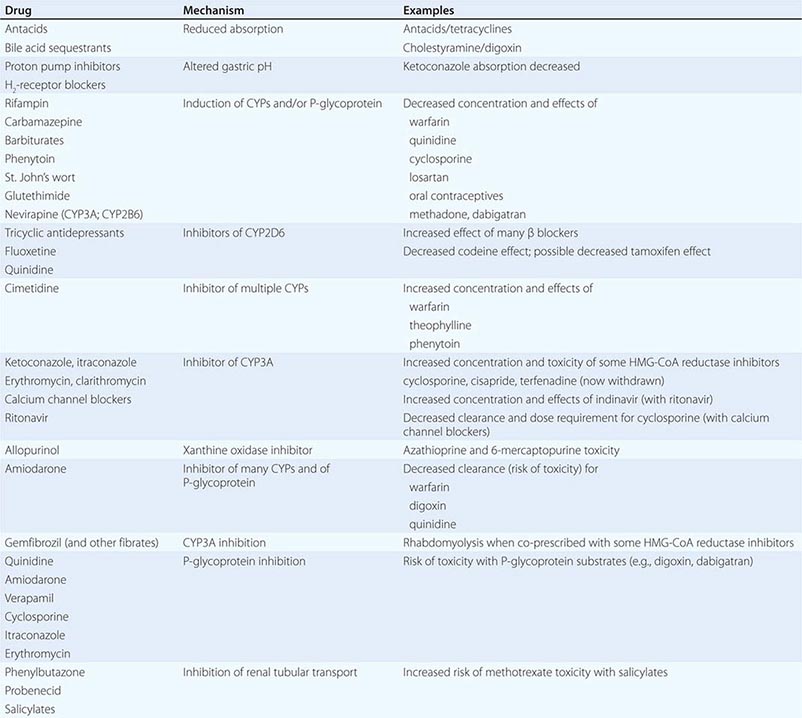
PHARMACOKINETIC INTERACTIONS CAUSING DECREASED DRUG EFFECTS
Gastrointestinal absorption can be reduced if a drug interaction results in drug binding in the gut, as with aluminum-containing antacids, kaolin-pectin suspensions, or bile acid sequestrants. Drugs such as histamine H2-receptor antagonists or proton pump inhibitors that alter gastric pH may decrease the solubility and hence absorption of weak bases such as ketoconazole.
Expression of some genes responsible for drug elimination, notably CYP3A and MDR1, can be markedly increased by inducing drugs, such as rifampin, carbamazepine, phenytoin, St. John’s wort, and glutethimide, and by smoking, exposure to chlorinated insecticides such as DDT (CYP1A2), and chronic alcohol ingestion. Administration of inducing agents lowers plasma levels, and thus effects, over 2–3 weeks as gene expression is increased. If a drug dose is stabilized in the presence of an inducer that is subsequently stopped, major toxicity can occur as clearance returns to preinduction levels and drug concentrations rise. Individuals vary in the extent to which drug metabolism can be induced, likely through genetic mechanisms.
Interactions that inhibit the bioactivation of prodrugs will decrease drug effects (Table 5-1).
Interactions that decrease drug delivery to intracellular sites of action can decrease drug effects: tricyclic antidepressants can blunt the antihypertensive effect of clonidine by decreasing its uptake into adrenergic neurons. Reduced CNS penetration of multiple HIV protease inhibitors (with the attendant risk of facilitating viral replication in a sanctuary site) appears attributable to P-glycoprotein-mediated exclusion of the drug from the CNS; indeed, inhibition of P-glycoprotein has been proposed as a therapeutic approach to enhance drug entry to the CNS (Fig. 5-5A).
PHARMACOKINETIC INTERACTIONS CAUSING INCREASED DRUG EFFECTS
The most common mechanism here is inhibition of drug elimination. In contrast to induction, new protein synthesis is not involved, and the effect develops as drug and any inhibitor metabolites accumulate (a function of their elimination half-lives). Since shared substrates of a single enzyme can compete for access to the active site of the protein, many CYP substrates can also be considered inhibitors. However, some drugs are especially potent as inhibitors (and occasionally may not even be substrates) of specific drug elimination pathways, and so it is in the use of these agents that clinicians must be most alert to the potential for interactions (Table 5-3). Commonly implicated interacting drugs of this type include amiodarone, cimetidine, erythromycin and some other macrolide antibiotics (clarithromycin but not azithromycin), ketoconazole and other azole antifungals, the antiretroviral agent ritonavir, and high concentrations of grapefruit juice (Table 5-3). The consequences of such interactions will depend on the drug whose elimination is being inhibited (see “The Concept of High-Risk Pharmacokinetics,” above). Examples include CYP3A inhibitors increasing the risk of cyclosporine toxicity or of rhabdomyolysis with some HMG-CoA reductase inhibitors (lovastatin, simvastatin, atorvastatin, but not pravastatin), and P-glycoprotein inhibitors increasing the risk of toxicity with digoxin therapy or of bleeding with the thrombin inhibitor dabigatran.
These interactions can occasionally be exploited to therapeutic benefit. The antiviral ritonavir is a very potent CYP3A4 inhibitor that is sometimes added to anti-HIV regimens, not because of its antiviral effects but because it decreases clearance, and hence increases efficacy, of other anti-HIV agents. Similarly, calcium channel blockers have been deliberately coadministered with cyclosporine to reduce its clearance and thus its maintenance dosage and cost.
Phenytoin, an inducer of many systems, including CYP3A, inhibits CYP2C9. CYP2C9 metabolism of losartan to its active metabolite is inhibited by phenytoin, with potential loss of antihypertensive effect.
Grapefruit (but not orange) juice inhibits CYP3A, especially at high doses; patients receiving drugs where even modest CYP3A inhibition may increase the risk of adverse effects (e.g., cyclosporine, some HMG-CoA reductase inhibitors) should therefore avoid grapefruit juice.
CYP2D6 is markedly inhibited by quinidine, a number of neuroleptic drugs (chlorpromazine and haloperidol), and the SSRIs fluoxetine and paroxetine. The clinical consequences of fluoxetine’s interaction with CYP2D6 substrates may not be apparent for weeks after the drug is started, because of its very long half-life and slow generation of a CYP2D6-inhibiting metabolite.
6-Mercaptopurine is metabolized not only by TPMT but also by xanthine oxidase. When allopurinol, an inhibitor of xanthine oxidase, is administered with standard doses of azathioprine or 6-mercaptopurine, life-threatening toxicity (bone marrow suppression) can result.
A number of drugs are secreted by the renal tubular transport systems for organic anions. Inhibition of these systems can cause excessive drug accumulation. Salicylate, for example, reduces the renal clearance of methotrexate, an interaction that may lead to methotrexate toxicity. Renal tubular secretion contributes substantially to the elimination of penicillin, which can be inhibited (to increase its therapeutic effect) by probenecid. Similarly, inhibition of the tubular cation transport system by cimetidine decreases the renal clearance of dofetilide.
DRUG INTERACTIONS NOT MEDIATED BY CHANGES IN DRUG DISPOSITION
Drugs may act on separate components of a common process to generate effects greater than either has alone. Antithrombotic therapy with combinations of antiplatelet agents (glycoprotein IIb/IIIa inhibitors, aspirin, clopidogrel) and anticoagulants (warfarin, heparins) is often used in the treatment of vascular disease, although such combinations carry an increased risk of bleeding.
Nonsteroidal anti-inflammatory drugs (NSAIDs) cause gastric ulcers, and in patients treated with warfarin, the risk of upper gastrointestinal bleeding is increased almost threefold by concomitant use of an NSAID.
Indomethacin, piroxicam, and probably other NSAIDs antagonize the antihypertensive effects of β-adrenergic receptor blockers, diuretics, ACE inhibitors, and other drugs. The resulting elevation in blood pressure ranges from trivial to severe. This effect is not seen with aspirin and sulindac but has been found with the cyclooxygenase 2 (COX-2) inhibitor celecoxib.
Torsades des pointes ventricular tachycardia during administration of QT-prolonging antiarrhythmics (quinidine, sotalol, dofetilide) occurs much more frequently in patients receiving diuretics, probably reflecting hypokalemia. In vitro, hypokalemia not only prolongs the QT interval in the absence of drug but also potentiates drug block of ion channels that results in QT prolongation. Also, some diuretics have direct electrophysiologic actions that prolong QT.
The administration of supplemental potassium leads to more frequent and more severe hyperkalemia when potassium elimination is reduced by concurrent treatment with ACE inhibitors, spironolactone, amiloride, or triamterene.
The pharmacologic effects of sildenafil result from inhibition of the phosphodiesterase type 5 isoform that inactivates cyclic guanosine monophosphate (GMP) in the vasculature. Nitroglycerin and related nitrates used to treat angina produce vasodilation by elevating cyclic GMP. Thus, coadministration of these nitrates with sildenafil can cause profound hypotension, which can be catastrophic in patients with coronary disease.
Sometimes, combining drugs can increase overall efficacy and/or reduce drug-specific toxicity. Such therapeutically useful interactions are described in chapters dealing with specific disease entities.
ADVERSE REACTIONS TO DRUGS
The beneficial effects of drugs are coupled with the inescapable risk of untoward effects. The morbidity and mortality from these adverse effects often present diagnostic problems because they can involve every organ and system of the body and may be mistaken for signs of underlying disease. As well, some surveys have suggested that drug therapy for a range of chronic conditions such as psychiatric disease or hypertension does not achieve its desired goal in up to half of treated patients; thus, the most common “adverse” drug effect may be failure of efficacy.
Adverse reactions can be classified in two broad groups. One type results from exaggeration of an intended pharmacologic action of the drug, such as increased bleeding with anticoagulants or bone marrow suppression with antineoplastics. The second type of adverse reaction ensues from toxic effects unrelated to the intended pharmacologic actions. The latter effects are often unanticipated (especially with new drugs) and frequently severe and may result from recognized as well as previously undescribed mechanisms.
Drugs may increase the frequency of an event that is common in a general population, and this may be especially difficult to recognize; an excellent example is the increase in myocardial infarctions with the COX-2 inhibitor rofecoxib. Drugs can also cause rare and serious adverse effects, such as hematologic abnormalities, arrhythmias, severe skin reactions, or hepatic or renal dysfunction. Prior to regulatory approval and marketing, new drugs are tested in relatively few patients who tend to be less sick and to have fewer concomitant diseases than those patients who subsequently receive the drug therapeutically. Because of the relatively small number of patients studied in clinical trials and the selected nature of these patients, rare adverse effects are generally not detected prior to a drug’s approval; indeed, if they are detected, the new drugs are generally not approved. Therefore, physicians need to be cautious in the prescription of new drugs and alert for the appearance of previously unrecognized adverse events.
Elucidating mechanisms underlying adverse drug effects can assist development of safer compounds or allow a patient subset at especially high risk to be excluded from drug exposure. National adverse reaction reporting systems, such as those operated by the FDA (suspected adverse reactions can be reported online at http://www.fda.gov/safety/medwatch/default.htm) and the Committee on Safety of Medicines in Great Britain, can prove useful. The publication or reporting of a newly recognized adverse reaction can in a short time stimulate many similar such reports of reactions that previously had gone unrecognized.
Occasionally, “adverse” effects may be exploited to develop an entirely new indication for a drug. Unwanted hair growth during minoxidil treatment of severely hypertensive patients led to development of the drug for hair growth. Sildenafil was initially developed as an antianginal, but its effects to alleviate erectile dysfunction not only led to a new drug indication but also to increased understanding of the role of type 5 phosphodiesterase in erectile tissue. These examples further reinforce the concept that prescribers must remain vigilant to the possibility that unusual symptoms may reflect unappreciated drug effects.
Some 25–50% of patients make errors in self-administration of prescribed medicines, and these errors can be responsible for adverse drug effects. Similarly, patients commit errors in taking OTC drugs by not reading or following the directions on the containers. Health care providers must recognize that providing directions with prescriptions does not always guarantee compliance.
In hospitals, drugs are administered in a controlled setting, and patient compliance is, in general, ensured. Errors may occur nevertheless—the wrong drug or dose may be given or the drug may be given to the wrong patient—and improved drug distribution and administration systems are addressing this problem.
SCOPE OF THE PROBLEM
Patients receive, on average, 10 different drugs during each hospitalization. The sicker the patient, the more drugs are given, and there is a corresponding increase in the likelihood of adverse drug reactions. When <6 different drugs are given to hospitalized patients, the probability of an adverse reaction is ~5%, but if >15 drugs are given, the probability is >40%. Retrospective analyses of ambulatory patients have revealed adverse drug effects in 20%. Serious adverse reactions are also well-recognized with “herbal” remedies and OTC compounds; examples include kava-associated hepatotoxicity, L-tryptophan-associated eosinophilia-myalgia, and phenylpropanolamine-associated stroke, each of which has caused fatalities.
A small group of widely used drugs accounts for a disproportionate number of reactions. Aspirin and other NSAIDs, analgesics, digoxin, anticoagulants, diuretics, antimicrobials, glucocorticoids, antineoplastics, and hypoglycemic agents account for 90% of reactions.
TOXICITY UNRELATED TO A DRUG’S PRIMARY PHARMACOLOGIC ACTIVITY
Drugs or more commonly reactive metabolites generated by CYPs can covalently bind to tissue macromolecules (such as proteins or DNA) to cause tissue toxicity. Because of the reactive nature of these metabolites, covalent binding often occurs close to the site of production, typically the liver.
The most common cause of drug-induced hepatotoxicity is acetaminophen overdosage (Chap. 361). Normally, reactive metabolites are detoxified by combining with hepatic glutathione. When glutathione becomes depleted, the metabolites bind instead to hepatic protein, with resultant hepatocyte damage. The hepatic necrosis produced by the ingestion of acetaminophen can be prevented or attenuated by the administration of substances such as N-acetylcysteine that reduce the binding of electrophilic metabolites to hepatic proteins. The risk of acetaminophen-related hepatic necrosis is increased in patients receiving drugs such as phenobarbital or phenytoin, which increase the rate of drug metabolism, or ethanol, which exhausts glutathione stores. Such toxicity has even occurred with therapeutic dosages, so patients at risk through these mechanisms should be warned.
Most pharmacologic agents are small molecules with low molecular weights (<2000) and thus are poor immunogens. Generation of an immune response to a drug therefore usually requires in vivo activation and covalent linkage to protein, carbohydrate, or nucleic acid.
Drug stimulation of antibody production may mediate tissue injury by several mechanisms. The antibody may attack the drug when the drug is covalently attached to a cell and thereby destroy the cell. This occurs in penicillin-induced hemolytic anemia. Antibody-drug-antigen complexes may be passively adsorbed by a bystander cell, which is then destroyed by activation of complement; this occurs in quinine- and quinidine-induced thrombocytopenia. Heparin-induced thrombocytopenia arises when antibodies against complexes of platelet factor 4 peptide and heparin generate immune complexes that activate platelets; thus, the thrombocytopenia is accompanied by “paradoxical” thrombosis and is treated with thrombin inhibitors. Drugs or their reactive metabolites may alter a host tissue, rendering it antigenic and eliciting autoantibodies. For example, hydralazine and procainamide (or their reactive metabolites) can chemically alter nuclear material, stimulating the formation of antinuclear antibodies and occasionally causing lupus erythematosus. Drug-induced pure red cell aplasia (Chap. 130) is due to an immune-based drug reaction.
Serum sickness (Chap. 376) results from the deposition of circulating drug-antibody complexes on endothelial surfaces. Complement activation occurs, chemotactic factors are generated locally, and an inflammatory response develops at the site of complex entrapment. Arthralgias, urticaria, lymphadenopathy, glomerulonephritis, or cerebritis may result. Foreign proteins (vaccines, streptokinase, therapeutic antibodies) and antibiotics are common causes. Many drugs, particularly antimicrobial agents, ACE inhibitors, and aspirin, can elicit anaphylaxis with production of IgE, which binds to mast cell membranes. Contact with a drug antigen initiates a series of biochemical events in the mast cell and results in the release of mediators that can produce the characteristic urticaria, wheezing, flushing, rhinorrhea, and (occasionally) hypotension.
Drugs may also elicit cell-mediated immune responses. Topically administered substances may interact with sulfhydryl or amino groups in the skin and react with sensitized lymphocytes to produce the rash characteristic of contact dermatitis. Other types of rashes may also result from the interaction of serum factors, drugs, and sensitized lymphocytes.
DIAGNOSIS AND TREATMENT OF ADVERSE DRUG REACTIONS
The manifestations of drug-induced diseases frequently resemble those of other diseases, and a given set of manifestations may be produced by different and dissimilar drugs. Recognition of the role of a drug or drugs in an illness depends on appreciation of the possible adverse reactions to drugs in any disease, on identification of the temporal relationship between drug administration and development of the illness, and on familiarity with the common manifestations of the drugs. A suspected adverse drug reaction developing after introduction of a new drug naturally implicates that drug; however, it is also important to remember that a drug interaction may be responsible. Thus, for example, a patient on a chronic stable warfarin dose may develop a bleeding complication after introduction of amiodarone; this does not reflect a direct reaction to amiodarone but rather its effect to inhibit warfarin metabolism. Many associations between particular drugs and specific reactions have been described, but there is always a “first time” for a novel association, and any drug should be suspected of causing an adverse effect if the clinical setting is appropriate.
Illness related to a drug’s intended pharmacologic action is often more easily recognized than illness attributable to immune or other mechanisms. For example, side effects such as cardiac arrhythmias in patients receiving digitalis, hypoglycemia in patients given insulin, or bleeding in patients receiving anticoagulants are more readily related to a specific drug than are symptoms such as fever or rash, which may be caused by many drugs or by other factors.
Electronic listings of adverse drug reactions can be useful. However, exhaustive compilations often provide little sense of perspective in terms of frequency and seriousness, which can vary considerably among patients.
Eliciting a drug history from each patient is important for diagnosis. Attention must be directed to OTC drugs and herbal preparations as well as to prescription drugs. Each type can be responsible for adverse drug effects, and adverse interactions may occur between OTC drugs and prescribed drugs. Loss of efficacy of oral contraceptives or cyclosporine with concurrent use of St. John’s wort (a P-glycoprotein inducer) is an example. In addition, it is common for patients to be cared for by several physicians, and duplicative, additive, antagonistic, or synergistic drug combinations may therefore be administered if the physicians are not aware of the patients’ drug histories. Every physician should determine what drugs a patient has been taking, for the previous month or two ideally, before prescribing any medications. Medications stopped for inefficacy or adverse effects should be documented to avoid pointless and potentially dangerous reexposure. A frequently overlooked source of additional drug exposure is topical therapy; for example, a patient complaining of bronchospasm may not mention that an ophthalmic beta blocker is being used unless specifically asked. A history of previous adverse drug effects in patients is common. Since these patients have shown a predisposition to drug-induced illnesses, such a history should dictate added caution in prescribing new drugs.
Laboratory studies may include demonstration of serum antibody in some persons with drug allergies involving cellular blood elements, as in agranulocytosis, hemolytic anemia, and thrombocytopenia. For example, both quinine and quinidine can produce platelet agglutination in vitro in the presence of complement and the serum from a patient who has developed thrombocytopenia following use of this drug. Biochemical abnormalities such as G6PD deficiency, serum pseudocholinesterase level, or genotyping may also be useful in diagnosis, often after an adverse effect has occurred in the patient or a family member.
Once an adverse reaction is suspected, discontinuation of the suspected drug followed by disappearance of the reaction is presumptive evidence of a drug-induced illness. Confirming evidence may be sought by cautiously reintroducing the drug and seeing if the reaction reappears. However, that should be done only if confirmation would be useful in the future management of the patient and if the attempt would not entail undue risk. With concentration-dependent adverse reactions, lowering the dosage may cause the reaction to disappear, and raising it may cause the reaction to reappear. When the reaction is thought to be allergic, however, readministration of the drug may be hazardous, since anaphylaxis may develop.
If the patient is receiving many drugs when an adverse reaction is suspected, the drugs likeliest to be responsible can usually be identified; this should include both potential culprit agents as well as drugs that alter their elimination. All drugs may be discontinued at once or, if this is not practical, discontinued one at a time, starting with the ones most suspect, and the patient observed for signs of improvement. The time needed for a concentration-dependent adverse effect to disappear depends on the time required for the concentration to fall below the range associated with the adverse effect; that, in turn, depends on the initial blood level and on the rate of elimination or metabolism of the drug. Adverse effects of drugs with long half-lives or those not directly related to serum concentration may take a considerable time to disappear.
SUMMARY
Modern clinical pharmacology aims to replace empiricism in the use of drugs with therapy based on in-depth understanding of factors that determine an individual’s response to drug treatment. Molecular pharmacology, pharmacokinetics, genetics, clinical trials, and the educated prescriber all contribute to this process. No drug response should ever be termed idiosyncratic; all responses have a mechanism whose understanding will help guide further therapy with that drug or successors. This rapidly expanding understanding of variability in drug actions makes the process of prescribing drugs increasingly daunting for the practitioner. However, fundamental principles should guide this process:
• The benefits of drug therapy, however defined, should always outweigh the risk.
• The smallest dosage necessary to produce the desired effect should be used.
• The number of medications and doses per day should be minimized.
• Although the literature is rapidly expanding, accessing it is becoming easier; electronic tools to search databases of literature and unbiased opinion will become increasingly commonplace.
• Genetics play a role in determining variability in drug response and may become a part of clinical practice.
• Electronic medical record and pharmacy systems will increasingly incorporate prescribing advice, such as indicated medications not used; unindicated medications being prescribed; and potential dosing errors, drug interactions, or genetically determined drug responses.
• Prescribers should be particularly wary when adding or stopping specific drugs that are especially liable to provoke interactions and adverse reactions.
• Prescribers should use only a limited number of drugs, with which they are thoroughly familiar.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


