Lymphocyte-depleted Hodgkin Lymphoma
C. Cameron Yin, MD, PhD
Key Facts
Clinical Issues
< 1% of cases of CHL
Lymph nodes: Retroperitoneal or abdominal > peripheral
Abdominal organs, bone marrow
B symptoms are frequent
Clinical stage III-IV disease
Current chemotherapy and radiation can cure disease in many patients
Microscopic Pathology
Lymph node architecture is usually diffusely effaced
Generalized depletion of small lymphocytes
Eosinophils, neutrophils, and plasma cells are usually scant or absent
± coagulative necrosis; ± sinusoidal invasion
± disordered nonbirefringent fibrillary fibrosis
3 morphologic variants
Diffuse fibrosis
Reticular or sarcoma-like
Mixed cellularity-like with numerous HRS cells
Ancillary Tests
CD30(+) in > 95%; CD15(+) in ˜ 70-80% of cases
pax-5(dim +) ˜ 90%), CD20(variably +) ˜ 20%
EBV(+) with latency type II pattern in subset of cases
CD45/LCA(-)
Top Differential Diagnoses
Nodular sclerosis Hodgkin lymphoma, grade 2
Anaplastic large cell lymphoma, either ALK1(+) or ALK1(-)
Nonhematopoietic neoplasms
Peripheral T-cell lymphoma
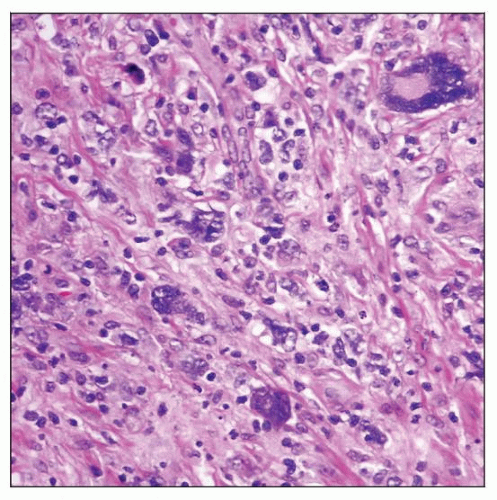 Lymphocyte-depleted Hodgkin lymphoma (LDHL) involving lymph node. Hodgkin and Reed-Sternberg (HRS) cells are numerous and highly pleomorphic, and small lymphocytes are depleted. |
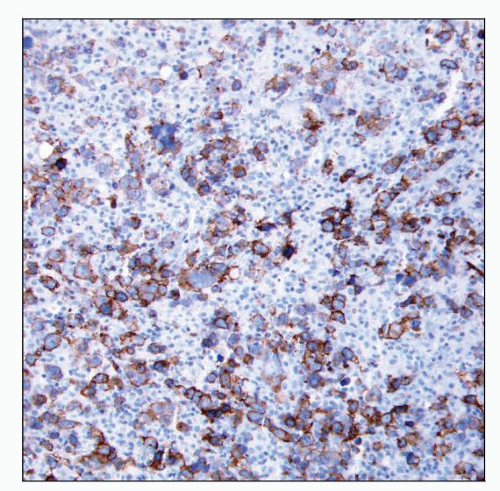 LDHL involving lymph node. Immunohistochemical analysis for CD30 highlights numerous HRS cells. Numerous HRS cells are a feature of the reticular morphologic variant of LDHL. |
TERMINOLOGY
Abbreviations
Lymphocyte-depleted Hodgkin lymphoma (LDHL)
Synonyms
Lymphocyte-depleted classical Hodgkin lymphoma
Lymphocyte-depleted (depletion) Hodgkin disease
Definitions
Classical Hodgkin lymphoma (CHL) is lymphoid neoplasm composed of Hodgkin and Reed-Sternberg (HRS) cells in variable inflammatory background
Lymphocyte depletion is a type of CHL characterized by depletion of small lymphocytes
Subset of cases has numerous &/or anaplastic HRS cells
ETIOLOGY/PATHOGENESIS
Infectious Agents
Epstein-Barr virus (EBV) probably has a pathogenic role in a subset of cases that are EBV(+)
HIV infection is associated with higher frequency of LDHL type
Pathogenesis
HRS cells arise from late germinal center or early post germinal center B cells that
Have undergone immunoglobulin (Ig) gene rearrangements with somatic mutations
Undergo crippling Ig gene mutations in subset of cases
Do not express B-cell antigen receptors
HRS cells lose much of normal B-cell immunophenotype due to
Severe impairment of transcription factor network regulating B-cell gene expression
Low or undetectable levels of transcription factors: OCT2, BOB1, PU.1, and early B-cell factor (EBF)
Leads to low level of Ig transcripts in HRS cells
Made worse by epigenetic silencing (promoter hypermethylation) of Ig transcription
Impaired function of early B cell development transcription factors: pax-5, E2A, and EBF
pax-5 dimly expressed or rarely absent in HRS cells
Aberrant overexpression of NOTCH1, ABF, and ID2 inhibit B-cell differentiation
Absent or dim expression of B-cell antigens: e.g., CD20
Overall these abnormalities physiologically should lead to apoptosis
However HRS are rescued from undergoing apoptosis
Development of antiapoptotic mechanisms to achieve survival
Inhibition of executors of apoptosis
Dysregulation of signaling pathways
Microenvironment is protective of HRS cells
LDHL most likely represents progression from other types of CHL
Suggested by older patient age at onset
CLINICAL ISSUES
Epidemiology
Incidence
< 1% of cases of CHL
Age
Median: 4th decade (or older in some studies)
Gender
M:F = 2-3:1
Site
Lymph nodes: Retroperitoneal or abdominal > peripheral
Abdominal organs, bone marrow
Presentation
B symptoms are frequent
Lymphadenopathy
Clinical stage III-IV disease
LDHL can spread contiguously (like other types of CHL) or by noncontiguous/vascular spread
Treatment
Chemotherapy ± radiation
Chemotherapy ABVD: Adriamycin (doxorubicin), bleomycin, vinblastine, and dacarbazine
Current chemotherapy and radiation can cure disease in many patients
Prognosis
Factors relevant to prognosis and to determination of mode of therapy
Male sex, B symptoms, high clinical stage
Elevated levels of serum LDH and β2-microglobulin
With therapy, prognosis of LDHL patients is similar to patients with other CHL types of similar stage
Recurrent disease with multiple adverse factors results in ˜ 60% overall survival at 5 years
MICROSCOPIC PATHOLOGY
Histologic Features
Lymph node architecture is usually diffusely effaced
Generalized depletion of small lymphocytes
Eosinophils, neutrophils, and plasma cells are usually scant or absent
± coagulative necrosis; ± sinusoidal invasion
± disordered nonbirefringent fibrillary fibrosis
3 morphologic variants
Diffuse fibrosis
Scant HRS cells admixed with few or abundant fibroblasts, fibrillary stroma, and scant lymphocytes
Reticular or sarcoma-like
Abundant HRS cells, including pleomorphic, bizarre (sarcomatous) cells
Capsular and perinodal infiltration are common
Mixed cellularity-like with numerous HRS cells
HRS cells include typical Reed-Sternberg cells and mononuclear variants
Cytologic Features
LDHL is difficult to diagnose in fine needle aspiration smears
Numerous HRS cells and depleted inflammatory background lead one away from diagnosis of CHL
ANCILLARY TESTS
Immunohistochemistry
CD30(+) in > 95%; CD15(+) in ˜ 70-80% of cases
Characteristic membranous pattern with accentuation in Golgi area
pax-5(dim +) ˜ 90%, CD20(variably +) ˜ 20%, CD79a(+) ˜ 10-20%
Ki-67(+), p53(+), MUM1(+)
CCL17(TARC)(+), fascin(+/-), Bcl-2(+/-)
CD45/LCA(-), EMA(-), Ig(-)
EBV(+) with latency type II pattern in ˜ 50% of cases
EBV-LMP(+), LMP2a(+), EBNA1(+), EBNA2(-)
Flow Cytometry
Polytypic B cells and T cells with normal immunophenotype
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


