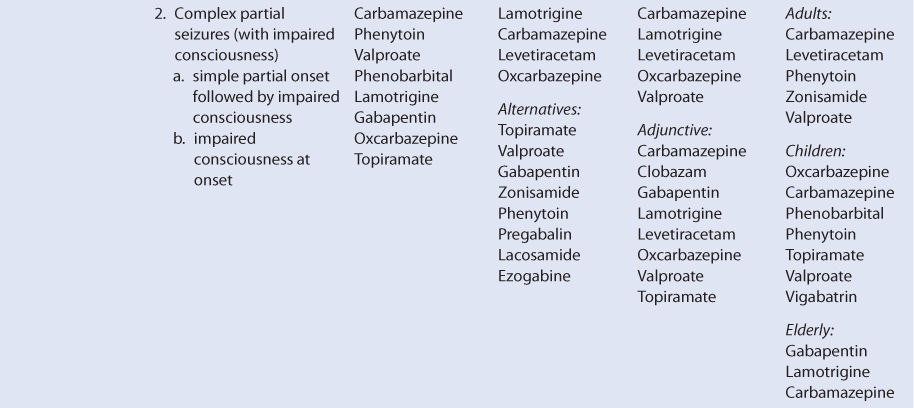
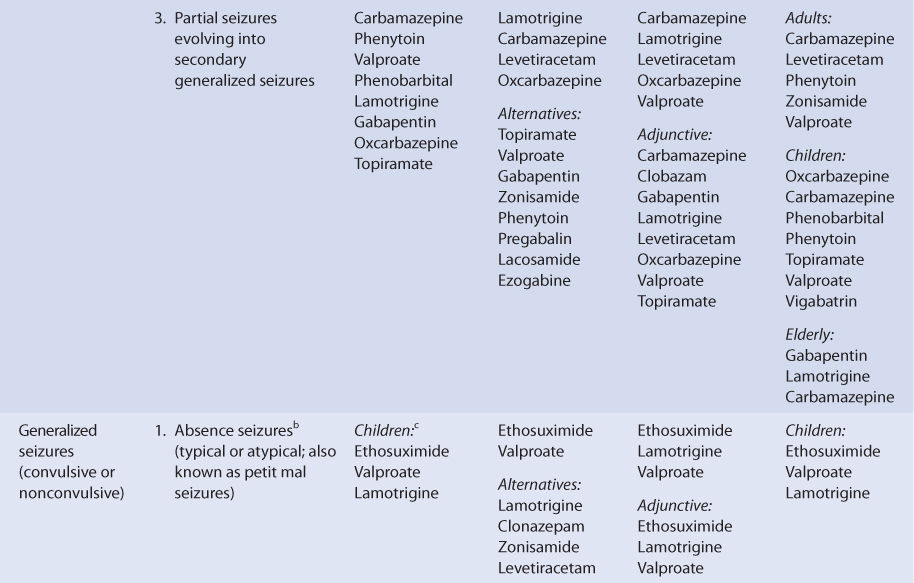

THERAPEUTIC AND TOXIC CONCENTRATIONS
Like many of the newer antiepileptic drugs, the therapeutic range of levetiracetam is not well defined.9–11 However, levetiracetam serum concentration measurement can be a useful adjunct in addition to monitoring treatment for therapeutic response and adverse effects. The therapeutic range for epilepsy used by many laboratories for levetiracetam is 12-46 μg/mL, and predose steady-state trough concentrations are usually obtained for levetiracetam measurement. Some patients will experience a higher incidence of adverse effects in the upper end of these levels, so the efficacy of lower serum concentrations should be assessed before moving to higher concentrations. The most common concentration-dependent side effects of levetiracetam are sedation and behavioral disturbances.6
CLINICAL MONITORING PARAMETERS
The goals of therapy with anticonvulsants are to reduce seizure frequency and to maximize quality of life with a minimum of adverse drug effects.6 While it is desirable to entirely abolish all seizure episodes, it may not be possible to accomplish this in many patients. Patients should be monitored for concentration-related side effects (sedation, behavioral disturbances). Somnolence, asthenia, and dizziness in adults and fatigue, aggression anorexia, and irritability in children are also common adverse events. Serious side effects include psychotic episodes (hallucinations, unusual behavior), mood changes (anxiety, depression, aggression, hostility), suicidal behavior, and muscle incoordination. Stevens-Johnson syndrome and toxic epidermal necrolysis have both been reported during levetiracetam therapy. Generally, these adverse effects occurred within 14-17 days of starting treatment, although some cases have happened as late as 4 months after beginning treatment. Should either of these dermatologic reactions develop, levetiracetam should be immediately discontinued and not restarted.6,12
Because epilepsy is an episodic disease state, patients do not experience seizures continuously. Thus, during dosage titration it is difficult to tell if the patient is responding to drug therapy or simply is not experiencing any abnormal central nervous system discharges at that time. Levetiracetam doses should be carefully titrated to individual patient response and tolerability. Steady-state serum concentrations of levetiracetam can be measured in patients as an adjunct to therapy.10,11,13–15 Concentration monitoring is particularly useful if a therapeutic level has been established for a patient and a new clinical event (drug interaction, new disease state or condition, etc) changes the pharmacokinetics of the drug. Levetiracetam concentrations can also be measured to document patient adherence to drug treatment or when a patient is switched among the various dosage forms (immediate-release tablets, extended-release tablets, oral solution, or intravenous injection) to assure therapeutic levels are maintained.
BASIC CLINICAL PHARMACOKINETIC PARAMETERS
With normal renal function, levetiracetam is eliminated primarily as unchanged drug in the urine (~65% of administered dose, Table 16-2). The remainder of the drug is largely converted to a pharmacologically inactive metabolite and then eliminated into the urine (UBC L057, ∼20% of dose). There are several other minor metabolites without antiseizure activity that account for the remainder of the dose. Metabolism of levetiracetam does not seem to be conducted principally by the cytochrome P-450 enzymes or the uridine 5′-diphosphoglucuronosyltransferase (UGT) systems. Enzymatic hydrolysis via type B esterases is a likely pathway for metabolite formation. The clearance, volume of distribution, and half-life for intravenous levetiracetam average about 0.95 mL/min/kg, 0.6 L/kg, and 7.3 hours, respectively. Levetiracetam follows linear pharmacokinetics within the usual dosage range.3,9,16–19
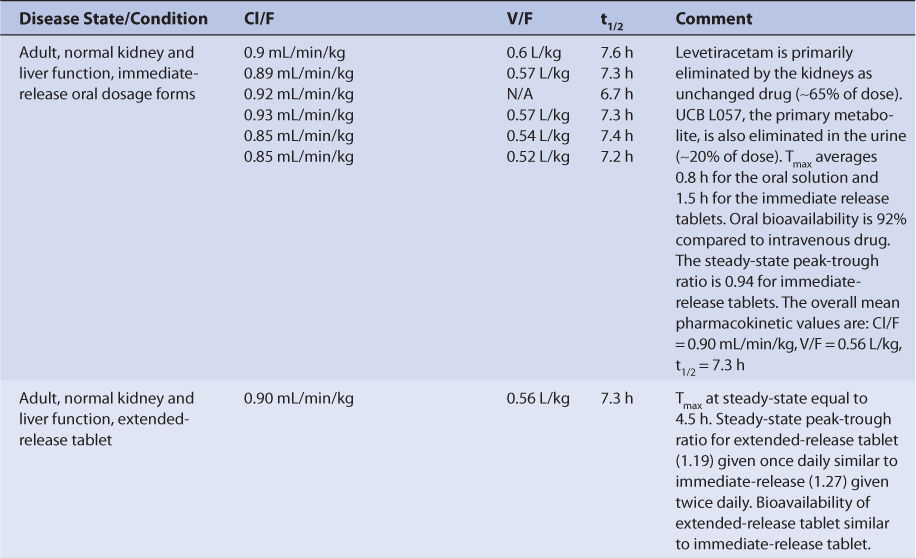
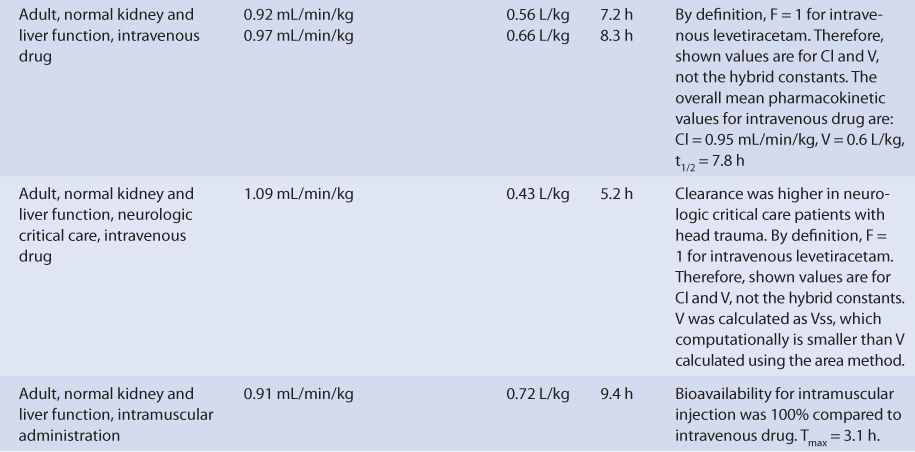
Levetiracetam is available in a variety of immediate-release tablets (tablets: 250 mg, 500 mg, 750 mg, 1000 mg) and as extended-release tablets (500 mg, 750 mg). An oral solution (100 mg/mL) and intravenous injection (500 mg/5 mL) are also available for administration. Bioavailability is ∼92% for the various oral dosage forms, and if the intravenous injection is given intramuscularly, the bioavailability is 100%. Food intake does not appear to alter bioavailability for the oral dosage forms.20,21
For single doses, peak concentrations occur sooner (Tmax) for the oral solution (∼0.8 hours) compared to the immediate-release tablets (∼1.5 hours).17,22–25 For multiple doses of the extended-release dosage form administered twice daily, Tmax occurs at about 4.5 hours. The steady-state maximum-to-minimum concentration ratio is 1.27 for immediate-release tablets given every 12 hours compared to 1.19 for extended-release tablets given once daily.26 When patients are switched between dosage forms, they should be closely monitored for changes in efficacy or toxicity, and measurement of levetiracetam serum concentrations should be considered before and after the change has been made.
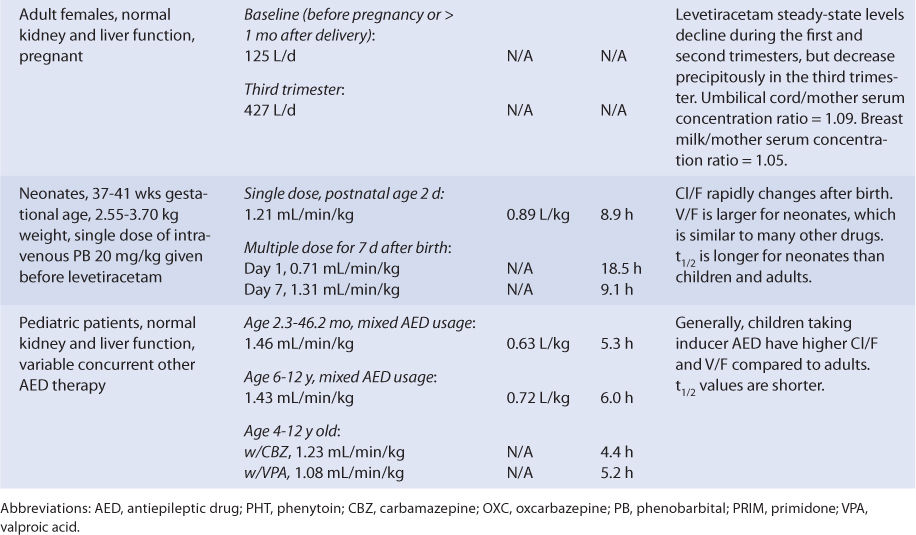
The plasma protein binding of levetiracetam is <10%.17–19 Levetiracetam crosses the placental barrier, and at birth the umbilical cord blood to maternal serum concentration ratio is approximately 1.09. Levetiracetam also crosses into the breast milk of lactating mothers with a breast milk-to-maternal serum ratio of 1.05. After breastfeeding from mothers taking levetiracetam, the resulting range of levetiracetam serum concentration in newborns was 0.7-3.4 μg/mL.13 The range of mean levetiracetam saliva to serum ratios in patients measured during multiple dosing is 0.36-0.41, and the correlation coefficient for concentrations in the two fluids is 0.86-0.87.27
EFFECTS OF DISEASES AND CONDITIONS ON PHARMACOKINETICS AND DOSING
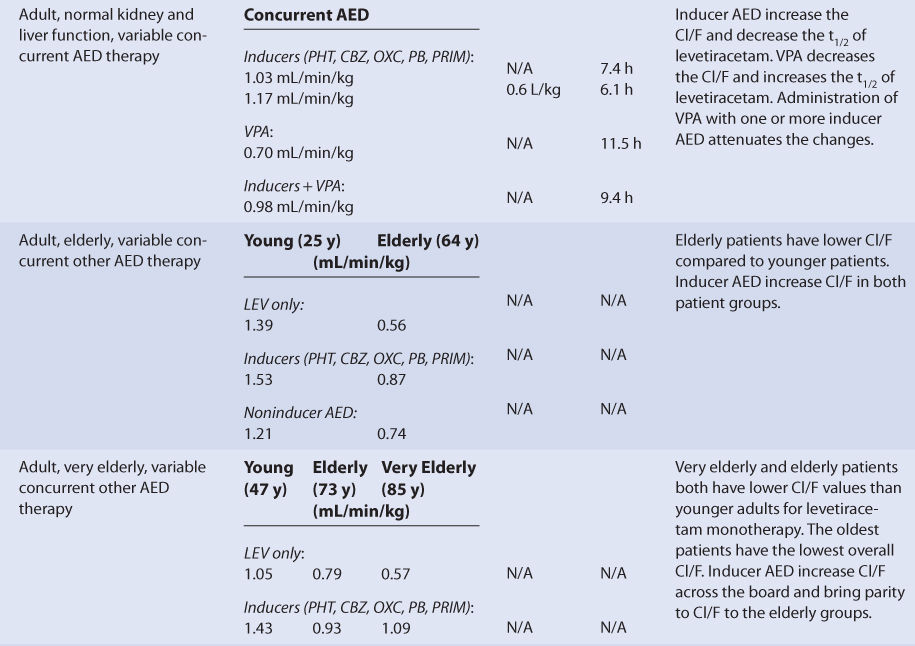
Most investigations with levetiracetam have been conducted with oral dosage forms, so pharmacokinetic results are usually expressed as hybrid constants that include bioavailability (F) for clearance (Cl/F) and volume of distribution (V/F) as single values (see Table 16-2). Adults with normal liver and kidney function that take no hepatic enzyme inducers or inhibitors have an average half-life of 7.3 hours, a mean Cl/F equal to 0.90 mL/min/kg, and an average V/F of 0.56 L/kg for levetiracetam (see Table 16-2).19,22–25 The clearance, volume of distribution, and half-life for intravenous levetiracetam average about 0.95 mL/min/kg, 0.6 L/kg, and 7.3 hours, respectively.20,21 Because plasma protein binding is very low (<10%), it is not altered by any disease states or conditions that alter levetiracetam pharmacokinetics, and volume of distribution is similar for most patient categories.
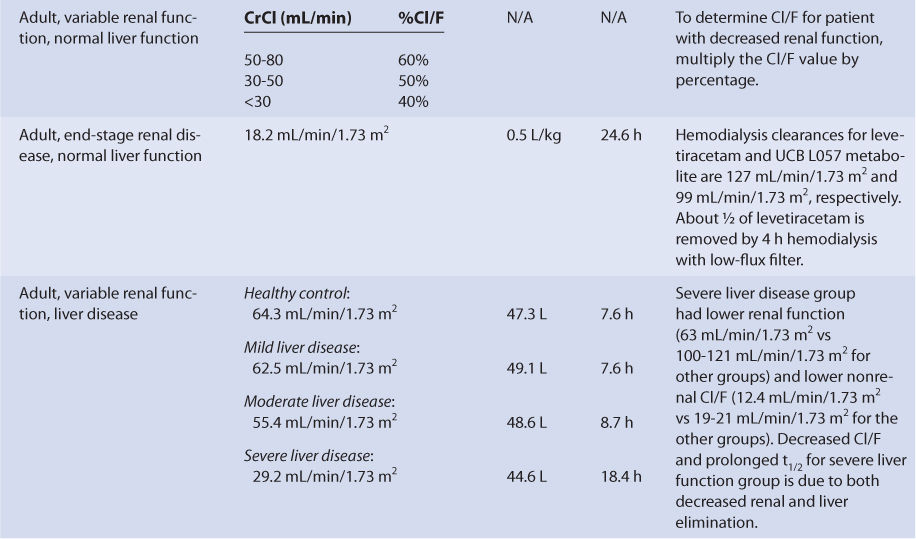
The majority of a levetiracetam dose (∼65%) is eliminated unchanged in the urine. Because of this, renal disease prolongs levetiracetam half-life and decreases levetiracetam clearance in a graded manner according to the degree of renal damage observed in the patient. Compared to adults with normal renal function, patients with a creatinine clearance (CrCl) of 50-80 mL/min have 60% of the normal Cl/F. Similarly, patients with CrCl equal to 30-50 mL/min have 50% of the normal Cl/F and patients with CrCl <30 mL/min have 40% of the normal Cl/F. Patients with end-stage renal disease have a levetiracetam clearance of 18.2 mL/min/1.73 m2, a volume of distribution equal to 0.5 L/kg, and a half-life of 24.6 hours. The hemodialysis clearances for levetiracetam and UCB L057 (the major metabolite) are 127 mL/min/1.73 m2 and 99 mL/min/1.73 m2, respectively. During a 4-hour hemodialysis session with a low-flux filter, about one-half of levetiracetam is removed from the body.28,29 For patients with renal dysfunction, levetiracetam doses should be titrated to clinical response, and measurement of levetiracetam serum concentrations can be a helpful adjunct. Due to the variability in the pharmacokinetics for levetiracetam, patients with renal disease should be closely monitored for adverse effects due to levetiracetam therapy.
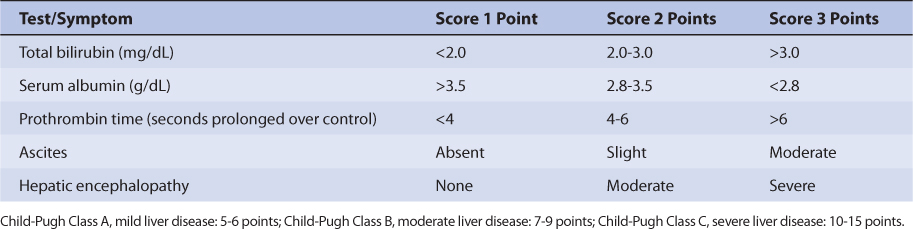
Since only a moderate amount of levetiracetam is eliminated by metabolic conversion (∼35%) and the liver is not the principal site of metabolism, there is only a modest change in levetiracetam pharmacokinetics for subjects with hepatic disease. An index of liver dysfunction can be gained by applying the Child-Pugh clinical classification system to the patient (Table 16-3).30 Child-Pugh scores are completely discussed in Chapter 3 (Drug Dosing in Special Populations: Renal and Hepatic Disease, Dialysis, Heart Failure, Obesity, and Drug Interactions), but will be briefly discussed here. The Child-Pugh score consists of five laboratory tests or clinical symptoms: serum albumin, total bilirubin, prothrombin time, ascites, and hepatic encephalopathy. Each of these areas is given a score of 1 (normal) to 3 (severely abnormal; see Table 16-3), and the scores for the five areas are summed. The Child-Pugh score for a patient with normal liver function is 5 while the score for a patient with grossly abnormal serum albumin, total bilirubin, and prothrombin time values in addition to severe ascites and hepatic encephalopathy is 15. The pharmacokinetic changes for levetiracetam range from modest ones (mild liver disease: none; moderate liver disease: half-life = 8.7 hours, Cl/F = 55.4 mL/min/1.73 m2, V/F = 48.6 L) for patients with moderate liver disease (Child-Pugh score = 7-9 points) to larger ones (half-life = 18.4 hours, Cl/F = 29.2 mL/min/1.73 m2) for patients with severe liver disease (Child-Pugh score = 10-15 points). Unfortunately, the patients with severe liver disease included in the research study also had decreased renal function compared to the other liver disease groups, so it is difficult to assign the entire change in levetiracetam pharmacokinetics to hepatic dysfunction. Generally, no change in dose is needed for patients with mild-moderate liver disease. But, for patients with severe liver disease (especially with a concurrent decline in renal function), an initial dosage decrease of 50% is recommended as a prudent change.31 Doses of levetiracetam for liver disease patients should be titrated to clinical response, and measurement of serum concentrations can be a helpful adjunct. Because there is variability in the pharmacokinetics for the agent, patients with liver disease should be closely monitored for adverse effects due to levetiracetam therapy.
Adult and pediatric patients taking levetiracetam for the treatment of seizures have been studied while taking other antiepileptic drugs, and it is important to take concurrent medications into account when interpreting the findings of pharmacokinetic studies (see Table 16-2). Because the metabolism of levetiracetam does not include the cytochrome P-450 system or the UGT enzymes, the induction (phenytoin, carbamazepine, oxcarbazepine, phenobarbital, primidone) or inhibition (valproic acid and its derivatives) effects of other antiepileptics is moderated to a large extent compared to other antiseizure medications. For adults, taking other antiepileptic drugs that are inducers simultaneously with levetiracetam will increase Cl/F by about 15%-30% and decrease half-life by about 16% for levetiracetam. By comparison, taking valproic acid or one of its derivatives that are inhibitors concurrently with levetiracetam will decrease Cl/F by ∼22% and increase half-life by ∼58% for levetiracetam. When either inducers or inhibitors are coadministered alone with levetiracetam, V/F for levetiracetam will remain unchanged with concomitant antiepileptic therapy. If antiepileptic drugs from both the inducer and the inhibitor categories are given at the same time with levetiracetam, the effect on levetiracetam pharmacokinetics are largely neutral (∼9% decrease in Cl/F, ∼29% increase in half-life).22,32 For the elderly and very elderly, the basic drug interaction pattern for levetiracetam with other antiepileptic drugs is the same, but adult patients over the age of 64 years have lower baseline levetiracetam Cl/F values for the effects to build upon.33,34
Pediatric patients taking other antiepileptic drugs that are inducers or inhibitors simultaneously with levetiracetam produces similar results for levetiracetam pharmacokinetic parameters. The inducer-class antiseizure medications (phenytoin, carbamazepine, oxcarbazepine, phenobarbital, primidone) increase Cl/F and decrease half-life for levetiracetam compared to patients taking levetiracetam monotherapy. The inhibitor-class antiseizure medications (valproic acid and its derivatives) decrease Cl/F and increase half-life for levetiracetam compared to patients taking levetiracetam monotherapy.35–37
Neonates (gestational age = 37-41 weeks, birth weight = 2550-3700 g) have been treated with intravenous levetiracetam to control seizures after delivery. Levetiracetam clearance on the first day of treatment was 0.71 mL/min/kg but rapidly increased over 7 days of therapy by 85%. During the same period, half-life decreased by 51% (see Table 16-2).
Intravenous levetiracetam has been investigated in normal individuals and patients with head trauma who were admitted to a neurologic critical care unit (see Table 16-2). Trauma patients are often hypermetabolic due to the physiologic demands placed on the body during the healing process. For this case, levetiracetam clearance is about 15% higher and half-life is about 33% shorter than normal adults.20,21,38 If the intravenous dosage form is administered intramuscularly to normal adults, Tmax averages 3.1 hours, and the bioavailability compared to intravenous drug is 100%.21
Using the FDA pregnancy classification system, levetiracetam is a Category C medication. Therefore, levetiracetam is used to treat pregnant women when the potential benefits of drug therapy outweigh the potential risks. Pregnancy alters the Cl/F of levetiracetam (see Table 16-2). Compared to baseline values, Cl/F continuously increases as the pregnancy progresses from the first to the second trimester, but a precipitous rise is noted during the third trimester. Clearance remains elevated until delivery, then starts to decline. By 1 month postpartum, levetiracetam clearance returns to baseline values. Levetiracetam crosses the placental barrier and is found in the breast milk of lactating mothers. At birth, the umbilical cord blood-to-maternal serum concentration ratio is approximately 1.09. The breast milk-to-maternal serum ratio is 1.05. After breastfeeding from mothers taking levetiracetam, the resulting range of levetiracetam serum concentration in newborns was 0.7-3.4 μg/mL.13
DRUG INTERACTIONS
Because the metabolism of levetiracetam does not involve the cytochrome P-450 system or the UGT enzymes, the induction (phenytoin, carbamazepine, oxcarbazepine, phenobarbital, primidone) or inhibition (valproic acid and its derivatives) effects of other antiepileptics on levetiracetam pharmacokinetics is moderate. Adult and pediatric patients taking levetiracetam for the treatment of seizures have been studied while taking other antiepileptic drugs, and it is important to take the effect of newly prescribed medications on levetiracetam pharmacokinetics into account when assessing a patient’s therapy (see Table 16-2).22,32–34 When other antiepileptic drugs are added to the treatment regimen of patients taking levetiracetam, patients should be monitored to ensure therapeutic effects are achieved and new adverse effects do not occur. Also, the effects of adding other common hepatic enzyme inducers or inhibitors to the therapeutic regimen of patients taking levetiracetam have not been thoroughly assessed. Measurement of levetiracetam steady-state concentrations can be a helpful adjunct when considering the effect of new therapies added to the medication regimen of epileptic patients.
INITIAL DOSAGE DETERMINATION METHODS
Several methods to initiate levetiracetam therapy are available. The Pharmacokinetic Dosing method is the most flexible of the techniques. It allows individualized target serum concentrations to be chosen for a patient, and each pharmacokinetic parameter can be customized to reflect specific disease states and conditions present in the patient. Literature-based recommended dosing is a very commonly used method to prescribe initial doses of levetiracetam. Doses are based on those that commonly produce steady-state concentrations in the lower end of the therapeutic range, although there is a wide variation in the actual concentrations for a specific patient (see Table 16-2).
Pharmacokinetic Dosing Method
The goal of initial dosing of levetiracetam is to compute the best dose possible for the patient given their set of disease states and conditions that influence levetiracetam pharmacokinetics and the epileptic disorder being treated. In order to do this, pharmacokinetic parameters for the patient will be estimated using average parameters measured in other patients with similar disease state and condition profiles.
Clearance, Volume of Distribution, and Half-Life Estimates
Levetiracetam is predominately eliminated unchanged by the kidney. Serum creatinine (SCr) and estimated creatinine clearance (CrClest) are used to estimate the elimination of agents that are renally eliminated, and there is a good correlation between creatinine clearance and levetiracetam clearance.29,39–41 Unfortunately, the exact equation that best describes the relationship between CrClest and levetiracetam clearance (Cl/F for orally administered drug, Cl for intravenously or intramuscularly administered drug) remains unpublished. Instead, clearance adjustments are used to account for decreased elimination for various CrClest brackets (see Table 16-2). Because of this, a patient is categorized according to the disease states and conditions that are known to change levetiracetam clearance, and the clearance previously measured in these studies is used as an estimate of the current patient’s levetiracetam clearance rate (see Table 16-2). Then, if the patient has a CrCl ≤80 mL/min, the clearance is adjusted for decreased renal function. For example, an adult epileptic patient with normal liver function that is to be initially treated with oral levetiracetam monotherapy (no other antiepileptic drug administration) would be assigned a levetiracetam Cl/F value equal to 0.90 mL/min/kg, a V/F value of 0.56 L/kg, and a t1/2 = 7.3 h.
If the patient has an estimated CrCl >80 mL/min, these estimates are used to compute levetiracetam dosages. If the patient has an estimated CrCl ≤80 mL/min, the clearance is adjusted for their renal function. To extend the current example, if the patient had a CrCl = 40 mL/min, the Cl/F value would be adjusted to 0.45 mL/min/kg (Cl/F = 0.50 • 0.90 mL/min/kg = 0.45 mL/min/kg), and the t1/2 value would be adjusted to 14.4 hours (t1/2 = [0.693(V/F)]/(Cl/F) = [0.693(0.56 L/kg)(1000 mL/L)]/[(0.45 mL/min/kg)(60 min/h)] = 14.4 h, where 1000 mL/L and 60 min/h are unit conversion factors). If the patient is receiving intravenous or intramuscular levetiracetam (F = 1), the same type of adjustment for renal function is made using Cl instead of Cl/F.
Selection of Appropriate Pharmacokinetic Model and Equations
When oral therapy is required, levetiracetam is usually given once daily for extended-release tablets or twice daily for the other oral dosage forms. These dosage intervals provide a relatively smooth serum concentration-time curve that approximates an intravenous infusion. Because of this concentration-time profile, a very simple pharmacokinetic equation that calculates the average levetiracetam steady-state concentration (Css in μg/mL = mg/L) is widely used and allows maintenance dose computation: Css = (D/τ)/(Cl/F) or D = Css(Cl/F)τ, where D is the dose of levetiracetam in mg, Cl/F is the levetiracetam hybrid clearance/bioavailability parameter in L/h, and τ is the dosage interval in hours.
If intravenous dosing is needed, levetiracetam is usually given twice daily as a 15-minute infusion. For this route of administration, a predose steady-state trough concentration (Cssmin) is usually maintained in the desired concentration range: Cssmin = (De−kτ)/[V(1 − e−kτ)] or D = [Cssmin V(1 − e−kτ)]/(e−kτ), where D is the dose of levetiracetam in mg, V is the volume of distribution in L, k is the elimination rate constant in h−1 [k = (0.693)/(t1/2)], and τ is the dosage interval in hours.

KL is a 50-year-old, 70-kg (height = 5 ft 10 in) male with complex partial seizures who requires therapy with levetiracetam. His serum creatinine equals 0.9 mg/dL. He has a normal liver function and is currently taking carbamazepine (steady-state concentration = 9.2 μg/mL). Additional dosage increases have been tried with carbamazepine, but they have been unsuccessful due to development of adverse effects. Suggest an initial levetiracetam dosage regimen using immediate-release tablets designed to achieve a steady-state levetiracetam concentration equal to 15 μg/mL.
1. Estimate creatinine clearance.
This patient has a stable serum creatinine and is not obese. The Cockcroft-Gault equation can be used to estimate creatinine clearance:
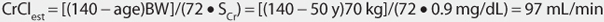
This patient has a CrClest >80 mL/min, so levetiracetam clearance will not need to be adjusted for renal function (see Table 16-2).
2. Estimate clearance, volume of distribution, and half-life according to disease states and conditions present in the patient.
The Cl/F rate for a patient taking carbamazepine is 1.17 mL/min/kg: Cl/F = (1.17 mL/min/kg)70 kg = 81.9 mL/min. The estimated V/F would be 0.6 L/kg, and the estimated half-life equals 6.1 h.
3. Compute dosage regimen.
Levetiracetam immediate-release tablets will be prescribed to this patient. (Note: μg/mL = mg/L and this concentration unit was substituted for Css in the calculations so that unnecessary unit conversion was not required.) The dosage equation for oral levetiracetam is: D = Css(Cl/F)τ = (15 mg/L • 81.9 mL/min • 12 h • 60 min/h)/(1000 mL/L) = 885 mg, rounded to 1000 mg every 12 hours.
A steady-state trough levetiracetam serum concentration can be measured after steady-state is attained in 3-5 half-lives. Since the patient is expected to have a half-life equal to 6.1 hours, the levetiracetam steady-state concentration could be obtained any time after the second day of dosing (5 half-lives = 5 • 6.1 h = 31 h or 1.3 d). Levetiracetam serum concentrations should also be measured if the patient experiences an exacerbation of their epilepsy or if the patient develops potential signs or symptoms of levetiracetam toxicity.
EXAMPLE 2 
Same patient profile as in example 1, but serum creatinine is 1.8 mg/dL indicating renal impairment.
1. Estimate creatinine clearance.
This patient has a stable serum creatinine and is not obese. The Cockcroft-Gault equation can be used to estimate creatinine clearance:
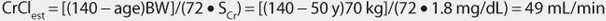
This patient has a CrClest = 30-50 mL/min, so levetiracetam clearance will be adjusted for renal function by multiplying Cl/F by 0.50 (see Table 16-2).
2. Estimate clearance, volume of distribution, and half-life according to disease states and conditions present in the patient.
The Cl/F rate for a patient taking carbamazepine is 1.17 mL/min/kg, and the adjustment for renal dysfunction is 0.50: Cl/F = (0.50)1.17 mL/min/kg = 0.59 mL/min/kg: Cl/F = (0.59 mL/min/kg)70 kg = 41.3 mL/min. The estimated V/F would be 0.6 L/kg, and the estimated half-life equals: t1/2 = [0.693(V/F)]/(Cl/F) = [0.693(0.6 L/kg)(1000 mL/L)]/[(0.59 mL/min/kg)(60 min/h)] = 11.7 h
Levetiracetam immediate-release tablets will be prescribed to this patient. (Note: μg/mL = mg/L and this concentration unit was substituted for Css in the calculations so that unnecessary unit conversion was not required.) The dosage equation for oral levetiracetam is: D = Css(Cl/F)τ = (15 mg/L • 41.3 mL/min • 12 h • 60 min/h)/(1000 mL/L) = 446 mg, rounded to 500 mg every 12 hours.
A steady-state trough levetiracetam serum concentration can be measured after steady-state is attained in 3-5 half-lives. Since the patient is expected to have a half-life equal to 11.7 hours, the levetiracetam steady-state concentration could be obtained any time after the third day of dosing (5 half-lives = 5 • 11.7 h = 59 h or 2.5 d). Levetiracetam serum concentrations should also be measured if the patient experiences an exacerbation of their epilepsy or if the patient develops potential signs or symptoms of levetiracetam toxicity.
EXAMPLE 3 
Same patient profile as in example 1, but patient not taking carbamazepine, and he will receive an intravenous levetiracetam dosage regimen designed to achieve a steady-state trough concentration equal to 15 μg/mL.
1. Estimate creatinine clearance.
This patient has a stable serum creatinine and is not obese. The Cockcroft-Gault equation can be used to estimate creatinine clearance:

This patient has a CrClest > 80 mL/min, so levetiracetam clearance will not need to be adjusted for renal function (see Table 16-2).
2. Estimate clearance, volume of distribution, and half-life according to disease states and conditions present in the patient.
The Cl for a patient taking intravenous levetiracetam as monotherapy is 0.95 mL/min/kg: Cl = (0.95 mL/min/kg) 70 kg = 66.5 mL/min. The estimated V would be 0.6 L/kg: V = (0.6 L/kg)70 kg = 42 L. The estimated half-life equals 7.8 h, so k = (0.693)/(t1/2) = (0.693)/(7.8 h) = 0.089 h−1.
3. Compute dosage regimen.
Levetiracetam intravenous injection will be prescribed to this patient. (Note: μg/mL = mg/L and this concentration unit was substituted for Css in the calculations so that unnecessary unit conversion was not required.) The dosage equation for intravenous levetiracetam is: D = [Cssmin V(1 − e−kτ)]/(e−kτ) = [15 mg/L • 42 L (1 − e−(0.089h−1)(12 h))]/(e−(0.089h−1)(12 h)) = 1203 mg, rounded to 1250 mg every 12 hours.
A steady-state trough levetiracetam serum concentration can be measured after steady-state is attained in 3-5 half-lives. Since the patient is expected to have a half-life equal to 7.8 hours, the levetiracetam steady-state concentration could be obtained any time after the second day of dosing (5 half-lives = 5 • 7.8 h = 39 h or 1.6 d). Levetiracetam serum concentrations should also be measured if the patient experiences an exacerbation of their epilepsy or if the patient develops potential signs or symptoms of levetiracetam toxicity.
Literature-Based Recommended Dosing
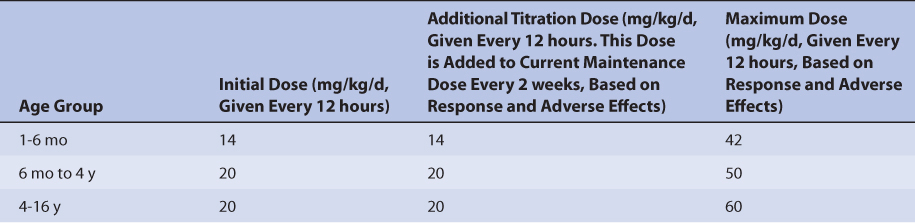
Because of the large amount of variability in levetiracetam pharmacokinetics, even when concurrent disease states and conditions are identified, most clinicians believe that the use of standard drug doses for various situations is warranted. The original computation of these doses were based on the Pharmacokinetic Dosing method, and subsequently modified based on clinical experience. In general, the expected steady-state serum concentrations used to compute these doses was in the lower end of the therapeutic range (12-15 μg/mL). For adults, the initial dose of levetiracetam is 1000 mg/d, administered once daily for the extended-release tablet and as divided doses every 12 hours for the other oral dosage forms. The dose is titrated up 1000 mg/d every 2 weeks as needed, and the patient is monitored for seizure frequency and adverse drug effects. The maximum dose is usually 3000-4000 mg/d. Because the majority of levetiracetam is eliminated unchanged in the urine, doses are adjusted for renal dysfunction (Table 16-4). If the patient has severe hepatic dysfunction (Child-Pugh score ≥10), the prescribed maintenance dose can be decreased by 50%. For children, the dose varies according to age, and Table 16-5 lists initial doses, dosage increases during the titration period, and the maximum recommended dose.42

TABLE 16-5 Literature-Based Initial Dose, Titration Rate, and Maximum Dose for Levetiracetam Immediate-Release Dosage Forms in Children
To illustrate the similarities and differences between this method of dosage calculation and the Pharmacokinetic Dosing method, the same examples used in the previous section will be used.
EXAMPLE 4 
KL is a 50-year-old, 70-kg (height = 5 ft 10 in) male with complex partial seizures who requires therapy with levetiracetam. His serum creatinine equals 0.9 mg/dL. He has normal liver function and is currently taking carbamazepine (steady-state concentration = 9.2 μg/mL). Additional dosage increases have been tried with carbamazepine, but they have been unsuccessful due to development of adverse effects. Suggest an initial levetiracetam dosage regimen using immediate-release tablets designed to achieve a steady-state levetiracetam concentration equal to 15 μg/mL.
1. Estimate creatinine clearance.
This patient has a stable serum creatinine and is not obese. The Cockcroft-Gault equation can be used to estimate creatinine clearance:
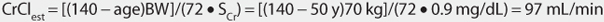
This patient has a CrClest > 80 mL/min, so levetiracetam clearance will not need to be adjusted for renal function (see Table 16-4).
2. Estimate levetiracetam dose according to disease states and conditions present in the patient.
For adults, the initial dose of levetiracetam is 1000 mg/d, administered as divided doses every 12 hours for immediate-release tablets. The dose is titrated up 1000 mg/d every 2 weeks as needed, and the patient is monitored for seizure frequency and adverse drug effects. The maximum dose is usually 3000-4000 mg/d.
A steady-state trough levetiracetam serum concentration can be measured after steady-state is attained in 3-5 half-lives. Since the patient is expected to have a half-life equal to 6.1 hours, the levetiracetam steady-state concentration could be obtained any time after the second day of dosing (5 half-lives = 5 • 6.1 h = 31 h or 1.3 d). Levetiracetam serum concentrations should also be measured if the patient experiences an exacerbation of their epilepsy or if the patient develops potential signs or symptoms of levetiracetam toxicity.
EXAMPLE 5 
Same patient profile as in example 1, but serum creatinine is 1.8 mg/dL indicating renal impairment.
1. Estimate creatinine clearance.
This patient has a stable serum creatinine and is not obese. The Cockcroft-Gault equation can be used to estimate creatinine clearance:
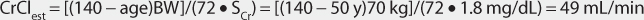
This patient has a CrClest between 30 and 50 mL/min, so levetiracetam clearance will be adjusted for renal function according to the recommendations in Table 16-4.
2. Estimate levetiracetam dose according to disease states and conditions present in the patient.
According to the guidelines given in Table 16-4, the initial dose for this patient should be 250 to 750 mg every 12 hours. Because the patient’s renal function is in the upper area of the CrClest adjustment range, the dose of 500 mg every 12 hours was prescribed.
A steady-state trough levetiracetam serum concentration can be measured after steady-state is attained in 3-5 half-lives. Since the patient is expected to have a half-life equal to 11.7 hours, the levetiracetam steady-state concentration could be obtained any time after the third day of dosing (5 half-lives = 5 • 11.7 h = 59 h or 2.5 d). Levetiracetam serum concentrations should also be measured if the patient experiences an exacerbation of their epilepsy or if the patient develops potential signs or symptoms of levetiracetam toxicity.
EXAMPLE 6 
Same patient profile as in example 1, but patient not taking carbamazepine, and he will receive an intravenous levetiracetam dosage regimen designed to achieve a steady-state trough concentration equal to 15 μg/mL.
1. Estimate creatinine clearance.
This patient has a stable serum creatinine and is not obese. The Cockcroft-Gault equation can be used to estimate creatinine clearance:
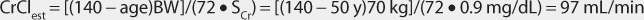
This patient has a CrClest >80 mL/min, so levetiracetam clearance will not need to be adjusted for renal function (see Table 16-2).
2. Estimate levetiracetam dose according to disease states and conditions present in the patient.
For adults, the initial dose of levetiracetam is 1000 mg/d, administered intravenously as divided doses every 12 hours. The drug is infused over 15 minutes. The dose is titrated up 1000 mg/d every 2 weeks as needed, and the patient is monitored for seizure frequency and adverse drug effects. The maximum dose is usually 3000-4000 mg/d.
A steady-state trough levetiracetam serum concentration can be measured after steady-state is attained in 3-5 half-lives. Since the patient is expected to have a half-life equal to 7.8 hours, the levetiracetam steady-state concentration could be obtained any time after the second day of dosing (5 half-lives = 5 • 7.8 h = 39 h or 1.6 d). Levetiracetam serum concentrations should also be measured if the patient experiences an exacerbation of their epilepsy or if the patient develops potential signs or symptoms of levetiracetam toxicity.
USE OF LEVETIRACETAM SERUM CONCENTRATIONS TO ALTER DOSES
A definitive therapeutic range for steady-state levetiracetam serum concentrations has not been established.9–11 Because of this, serum concentration monitoring for levetiracetam plays only a supportive role in the dosing of the drug. Important patient parameters (seizure frequency, potential levetiracetam side effects, etc) should be followed to confirm that the patient is responding to treatment and not developing adverse drug reactions.6 However, there are clinical situations where levetiracetam serum concentrations can be very helpful.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


