Chapter Fifty-Two. Jaundice and common metabolic problems in neonates
Neonatal jaundice
Morphological factors
Details of the structure and function of the mature liver are given in Chapter 22. As hepatic immaturity, hepatic disease and jaundice are common in neonates it is important to understand the morphological features and functional scope of the liver and its biliary tract. The liver is one of the largest visceral organs in neonates and normally extends from the right hypochondrium, through the epigastrium into the left hypochondrium, descending 2.5–3 cm below the costal margin. As an outgrowth of the caudal part of the foregut, the liver retains its connection with the gastrointestinal system via the biliary tree. In mature livers the hepatocytes are arranged into plates which are usually one cell thick (until the age of 7 years these plates are two cells thick) and separated by venous sinusoids which anastomose with each other forming larger veins which join corresponding central veins. The hepatic parenchyma is supplied by nerves arising from the hepatic plexus and containing sympathetic and parasympathetic (vagal) fibres. Distension or disruption of the liver capsule may cause localised sharp pain (Borley et al 2008).
A complex network of systemic and portal vessels ensures that the liver benefits from good blood supply and venous and lymphatic drainage. The portal venous system ensures that most of the blood from the gastrointestinal system is returned to the liver. In contrast, the hepatic venous system returns blood from the liver into the inferior vena cava. The protein-rich lymph is drained from the liver into the thoracic duct.
Synthesis and metabolism of bilirubin
Small amounts of bilirubin are detected in the amniotic fluid between the 13th and 37th weeks of gestation, possibly as a consequence of the limitations of the immature fetal liver to remove and conjugate it. This implies that the major route for fetal bilirubin excretion is across the placenta. Because most of the fetal plasma bilirubin is unconjugated it is easily transferred across the placenta into the maternal circulation and to the maternal liver for conjugation and excretion. Therefore fetuses seldom manifest jaundice unless they suffer severe haemolytic disease.
In neonates, 75% of bilirubin is a by-product of haemoglobin of which 1 g yields 35 mg of bilirubin (Frank & Frank 2006) (Fig. 52.1). About 25% is generated from other non-erythropoietic haem protein sources, principally in the liver and the destruction of immature erythrocytes in the bone marrow. Senescent erythrocytes are removed and destroyed by the reticuloendothelial system where haemoglobin is catabolised and converted into bilirubin. The iron atoms at the centre of the haem skeleton are reused. On leaving the reticuloendothelial system unconjugated bilirubin is transported in the plasma tightly bound to albumin. Although hepatocytes easily take up the unbound lipid-soluble bilirubin a carrier molecule is needed to take up any bound complexes. Once in the hepatocytes, bilirubin binds to ligandin and possibly other cytoplasmic binding proteins. The unconjugated bilirubin is conjugated with a sugar/glucuronic acid by enzymes to form bilirubin monoglucuronide and diglucuronide pigments which are more water-soluble and can be excreted into the bile or through the kidneys into the urine (Fig. 52.2). Bilirubin conjugation requires oxygen and glucose, and hypoxia or hypoglycaemia may slow down the conjugation process.
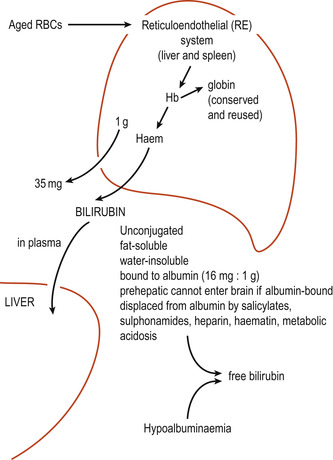 |
| Figure 52.1 The formation of bilirubin. (From Kelnar C, Harvey D, Simpson C 1995, with permission.) |
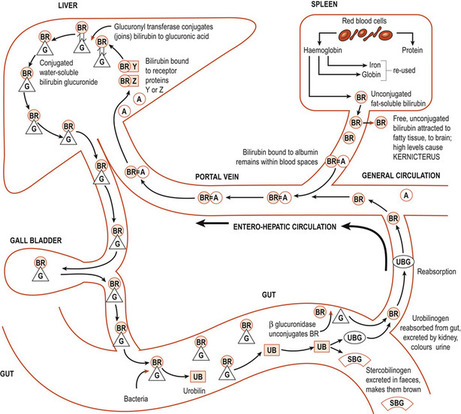 |
| Figure 52.2 Schematic diagram showing the conjugation of bilirubin. BR, bilirubin; =, bound to; A, albumin; G, glucuronic acid; UB, urobilin; UBG, urobilinogen; SBG, stercobilinogen. (From Bennett VR, Brown L 1993, with permission.) |
Some of the conjugated bilirubin is carried in bile to the duodenum and metabolised in the terminal ileum and colon by bacterial enzymes to produce urobilinogen ( stercobilinogen in the gut), which gives faeces the yellowish colour in infants and brown colour in adults. In its absence faeces are greyish white. Small quantities of urobilinogen re-enter the circulation and are excreted by the liver and the kidneys as hydrophilic urobilinogen.
Bilirubin in the neonate
After birth the neonate’s liver must take over bilirubin metabolism and clearance. Most of this bilirubin is diverted into the meconium; 200 g of meconium contains about 175 g of bilirubin, 50% of which is conjugated. Delays in meconium excretion, which occur in premature neonates and neonates with cystic fibrosis, may contribute to the return of some unconjugated bilirubin into the systemic circulation.
Term neonates hold about 137–171 μmol of bilirubin per litre of plasma (Maisels 1999). These high values compared to bilirubin values in adults are attributed to higher circulating erythrocytes at birth which are gradually biodegraded. The shorter erythrocyte life span of 80 days, as opposed to 120 days in adults, and a corresponding increase in immature or fragile erythrocytes means that more erythrocytes must be degraded (Madan et al 2005). The less-effective bilirubin binding to albumin, enhanced absorption of bilirubin through the enterohepatic circulation and the somewhat dormant hepatic conjugating system further complicate bilirubin conjugation and excretion. All neonates manifest a progressive rise in unconjugated bilirubin, which peaks at 180 μmol/L on the 3rd or 4th day of life, giving rise to physiological jaundice. The increase in unconjugated bilirubin, a relatively low liver uptake of bilirubin and the transient deficiency of the glucuronyl transferase enzyme culminate in reduced excretion of conjugated bilirubin. Although healthy neonates produce almost twice as much bilirubin as adults, their plasma bilirubin values fall to physiological norms within 6 weeks of birth.
Whilst the mechanisms involved in bilirubin conjugation are not fully understood, evidence shows that bilirubin is synthesised by, and released from, the reticuloendothelial system into the plasma and binds to plasma albumin. The higher levels of unconjugated bilirubin in neonates are attributed to lower plasma albumin values and lower albumin affinity for bilirubin. This is possibly due to the undeveloped molecular structure of the albumin (Blackburn 2007) and that some albumin-binding sites may be occupied by other molecular substances such as drugs, including heparin and chloramphenicol (Maisels 1999).
Kernicterus
Although bilirubin plays important physiological roles as an antioxidant and a free radical scavenger, its tendency to aggregate increases the risks of neurotoxicity. Plasma pH affects significantly the solubility of bilirubin and its binding to tissue sites: solubility decreases as plasma pH falls. Bilirubin may bind to cell membranes, compromise Na +–K + exchange, lower cell membrane potential, reduce mitochondrial viability, augment the basal ganglia and decrease activity in the auditory brain stem (Cashor 1990, cited by Maisels 1999). Bilirubin is preferentially deposited in the basal ganglia; Maisels (1999) suggests this may be because bilirubin attaches first to nerve terminals where it may lower membrane potentials. This may decrease nerve conduction and allow a retrograde uptake of bilirubin into cell bodies where it binds with various cellular components, staining these and augmenting their function.
Delays in bilirubin conjugation are of concern as unbound, unconjugated fat-soluble bilirubin deposits in neural tissue and visceral organs. Although under normal circumstances bilirubin flows constantly into and out of the brain, it is difficult under experimental conditions to keep bilirubin in the brain of ‘healthy animals’ or produce significant electrophysiological changes. However, asphyxiated animals and those who have a defective blood–brain barrier experienced ‘gross staining of the brain and electrophysiological changes’ (Maisels 1999). An intact blood–brain barrier probably excludes most water-soluble substances and protein but is permeable to lipid-soluble substances that are not protein-bound. Large proteins such as albumin are excluded from the brain but may cross the blood–brain barrier when its impermeability is compromised.
However, modest elevation of unconjugated serum bilirubin can produce reversible clinical and electrophysiological alterations in healthy full-term infants (Bratlid 1990). The second mechanism for bilirubin entry into the brain occurs when there is a marked increase in unbound bilirubin. Acidosis may facilitate the transfer of bilirubin across the blood–brain barrier and its subsequent deposition in the neural tissue of the brain (Maisels 1999). Although neural mitochondria contain the enzyme bilirubin oxidase, which converts bilirubin to biliverdin and other non-toxic products, the risk of irreversible brain damage caused by unconjugated bilirubin is high.
The risk of bilirubin encephalopathy (occuring in neonates with mild, reversible hyperbilirubinaemia) is increased in low-birth-weight neonates and those who experience hypoxic ischaemic insults. However, autopsy reports on jaundiced neonates also reveal bilirubin staining of the aorta, pleural fluid and visceral organs (Maisels 1999). In the brain unconjugated bilirubin leads to irreversible staining, especially in the basal ganglia and cerebellum, and causes kernicterus (yellow kernel). Deposition of larger quantities of unconjugated bilirubin may cause irreversible neural damage with long-term sequelae of cerebral athetosis and significant mental retardation. Such events are most likely to occur when the neonate’s serum bilirubin level rises above 350 μmol/L although concerns are raised when the bilirubin level rises above 250 μmol/L. Kernicterus is preventable in all but very small, sick neonates where the blood–brain barrier may be ineffective and quite low levels of bilirubin may damage the fragile tissues.
A résumé of common causes of neonatal jaundice
Jaundice (yellow discoloration of the skin and sclera caused by deposits of conjugated and unconjugated bilirubin) becomes visible when the serum bilirubin rises above 85 μmol/L. Bilirubin is a yellowish-green bile pigment which, prior to conjugation, is a weak acid. It is very soluble in lipid but only slightly soluble in water, making its excretion difficult. About 75% of bilirubin is released along with biliverdin when red blood cells are broken down in the liver and spleen, releasing haem and globin to the body stores. The remainder is derived from other haem products. Excessive bilirubin may enter the bloodstream in three ways (McCance & Huether 2006), all of which may affect the neonate:
1. During excessive haemolysis of red blood cells (prehepatic).
2. Where liver disease affects the metabolism and excretion of bile (hepatic).
3. Congenital obstruction of a component of the biliary system, e.g. congenital biliary atresia (post-hepatic).
As each of the above problems requires different therapeutic interventions, an appropriate range of investigations is needed if the:
• Jaundice appears within the first 24 h after birth.
• Jaundice persists for longer than 2 weeks.
• Neonate’s total bilirubin level is >250 μmol/L.
• Conjugated hyperbilirubinaemia is >300 μmol/L.
• Jaundice appears in an ill baby irrespective of its age and fails to respond to treatment.
It is important to distinguish between unconjugated and conjugated forms of bilirubin. The common causes of neonatal jaundice are shown in Table 52.1; Figure 52.3 illustrates the sites of events leading to jaundice.
| Prehepatic: unconjugated bilirubin | Hepatic: unconjugated bilirubin | Hepatic: mixed unconjugated and conjugated bilirubin | Posthepatic: unconjugated mixed and conjugated bilirubin | Posthepatic: unconjugated bilirubin |
|---|---|---|---|---|
| Physiological jaundice/jaundice of prematurity Haemolytic jaundice Bruising and haematoma Polycythaemia Postnatal infections | Breast milk jaundice | Congenital hypothyroidism Inborn errors of metabolism: galactosaemia Hepatitis, due to transplacental infections (TORCH organisms) | Congenital biliary atresia Bile plug syndrome | Paralytic ileus High intestinal obstruction such as duodenal atresia |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


