- Isoniazid inhibits the synthesis of mycolic acids, which are the building blocks for the mycobacterial cell wall.
- Isoniazid is used in combination with other agents to treat active tuberculosis infection; it is included in both the initial phase and continuation phase of therapy.
- Isoniazid is also used as monotherapy to prevent the development of active tuberculosis from latent infection.
Isoniazid (INH) (Figure 5.4.1) is a key component of the combination therapy used to treat tuberculosis (TB) and latent TB.
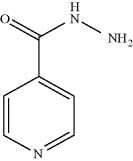
5.4.1 Discovery
As is the case with a number of drugs, most notably azidothymidine (AZT), a nucleoside reverse transcriptase inhibitor (NRTI) used in the treatment of HIV infections, isoniazid was first synthesised many years before the discovery of the antimycobacterial activity which led to its use in the treatment of TB. Isoniazid (isonicotinic acid hydrazide, INH) was originally synthesised in 1912 as part of a purely organic chemistry investigation (Meyer and Mally, 1912) and it was not until 1952 that its potential in the treatment of the dreaded TB (or consumption) was discovered (Wang, 2005).
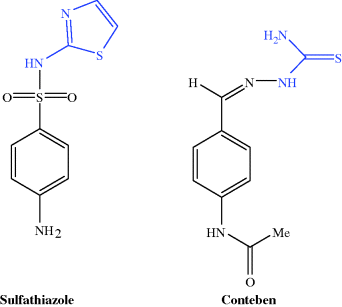
Interestingly, the discovery of isoniazid was partly due to a Nobel Prize winner we have already encountered in Section 3.1, Gerhard Domagk, who had been screening compounds against Mycobacterium tuberculosis (the organism responsible for TB) in the drug-discovery programme which resulted in the sulfonamides. A sulfonamide, sulfathiazole, had been shown to have weak activity against tubercule bacilli and a series of analogues was prepared, with the greatest activity found for compounds which were precursors of the thiazole ring, the thiosemicarbazones, and this eventually led to thiacetazone (Tb 1/698), later named conteben (Figure 5.4.2).
Figure 5.4.2 Sulfathiazole and conteben (thiacetazone), showing the similarity of the thiazolylamine and thiosemicarbazone portions
News of the trials of conteben in Germany in the late 1940s was soon picked up in the rest of the world and H. Corwin Hinshaw (Stanford University) and Walsh McDermott (Cornell Medical School) visited Europe to observe the effects of this drug on TB patients. The Americans returned home with a sample of conteben, and an amazing series of coincidences resulted in four independent laboratories working on isoniazid at the same time – at Roche and Squibb in the USA, Wellcome in the UK, and Domagk in Germany, all of whom thought that the intellectual property belonged to them! Having been synthesised 40 years previously, isoniazid was actually free of any patent restrictions and production of isoniazid (brand name, Pycazide) began in the UK in 1951 at Herts Pharmaceuticals Ltd. The worldwide interest in the 1950s in nicotinamide/isoniazid analogues for the treatment of TB led to other agents, including pyrazinamide and ethionamide, both of which are still used in some combination-treatment regimens (Greenwood, 2008).
5.4.2 Synthesis
As mentioned earlier, isoniazid was first synthesised in 1912, from isonicotinic acid ethyl ester and hydrazine (Meyer and Mally, 1912), and recent synthetic studies have concentrated on the use of more readily available starting materials such a 4-cyanopyridine (Scheme 5.4.1) (Sycheva et al., 1971).
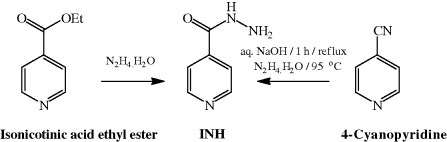
5.4.3 Bioavailability
Isoniazid has excellent bioavailability after oral administration as a single agent, at around 100% absorption, with peak blood concentrations achieved after 0.9–1 hours, peak mean serum concentrations of 3.6 μg/mL, and a half-life of 1.9 hours (Peters, 1960; Ellard and Gammon, 1976). As isoniazid is used as one component of combination therapy for TB, the pharmacokinetic parameters have also been investigated as part of a combination therapy; fortunately, they do not differ significantly from the single-agent parameters (Zwolska et al., 2002).
The absorption of isoniazid is affected by the presence of food (Melander et al., 1976) and by aluminum hydroxide (Hurwitz and Schlozman, 1974), with the result that it should not be taken with food or less than 1 hour before antacid formulations. TB is a complicating factor for many patients with AIDS, some of whom are already receiving didanosine, which incorporates antacid into the formulation to reduce stomach acidity and minimise the acid-catalysed hydrolysis of didanosine; fortunately, the small amount of antacid in the didanosine formulation has little effect upon the absorption of isoniazid, allowing its concurrent administration (Gallicano et al., 1994).
The bioavailability of isoniazid, like that of the sulfonamides, is affected by the rate at which it is metabolised by the human cytochrome enzyme, N-acetyltransferase 2 (NAT2) (Hughes, 1954; Evans et al., 1960). A patient with rapid acetylation of isoniazid metabolises and excretes it at a faster rate than is appropriate for the antibacterial activity, with the result that sub-therapeutic doses are achieved, with associated poor therapeutic outcomes. The situation is little better for those with slow acetylation status, as there is increased opportunity for high serum concentrations to be achieved, due to decreased metabolism and excretion of the isoniazid, and adverse events are more common for these patients; you will discover this in more detail in Subsection 5.4.7. As acetylator status is inherited and can be determined before treatment by genotype analysis, the personalisation of isoniazid treatment according to NAT2 status has been considered, and this would help to avoid adverse events caused by this problem (Evans, 1989; Spielberg, 1996; Butcher et al., 2002). When genotype analysis becomes less expensive and more routine, this may be a development for the future (Kinzig-Schippers et al., 2005).
5.4.4 Mode of Action and Selectivity
As mentioned in Section 1, mycobacteria are aerobic organisms, characterised by thicker cell walls than Gram positive and negative organisms. The mycobacterial cell wall is hydrophobic/waxy in nature, being rich in very-long-chain fatty acids, known as mycolic acids, and is resistant to agents which interfere with cell wall synthesis, such as β-lactams. Isoniazid is actually a prodrug and is converted in vivo to the active form by the mycobacterial enzyme catalase-peroxidase (KatG) (Zhou et al., 2006). Catalase-peroxidases protect bacteria from the cellular damage caused by reactive oxygen species and KatG is responsible for the high resistance of M. tuberculosis to hydrogen peroxide (Manca et al., 1999).
KatG is believed to activate isoniazid to the highly reactive isonicotinoyl radical, which complexes with the coenzymes NAD+or NADP+ (bound in the active site of InhA) to give an INH-NAD(P) adduct (Scheme 5.4.2), in which the isonicotinoyl group has replaced the 4S-hydrogen involved in the hydride transfer reduction of enoyl-ACP substrates (Rozwarski et al., 1998; Cade et al., 2010).
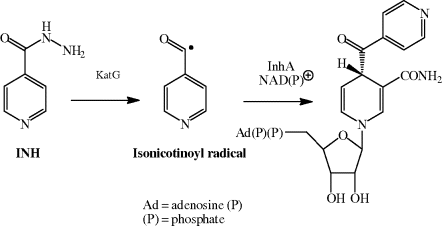
This adduct inhibits InhA, an NADH-dependent enoyl-acyl carrier protein reductase which catalyses the reduction of 2,3-trans-enoylACP through competition with the NADH enzyme cofactor (Scheme 5.4.3 and Figure 5.4.3) (Argyrou et al., 2007). InhA preferentially reduces long-chain fatty acid precursors to give products that are utilised by the mycobacterial type II fatty acid synthase system (FAS-II) to produce the very-long-chained α-branched mycolic acids, which are key components of the mycobacterial cell wall (Rozwarski et al., 1998).
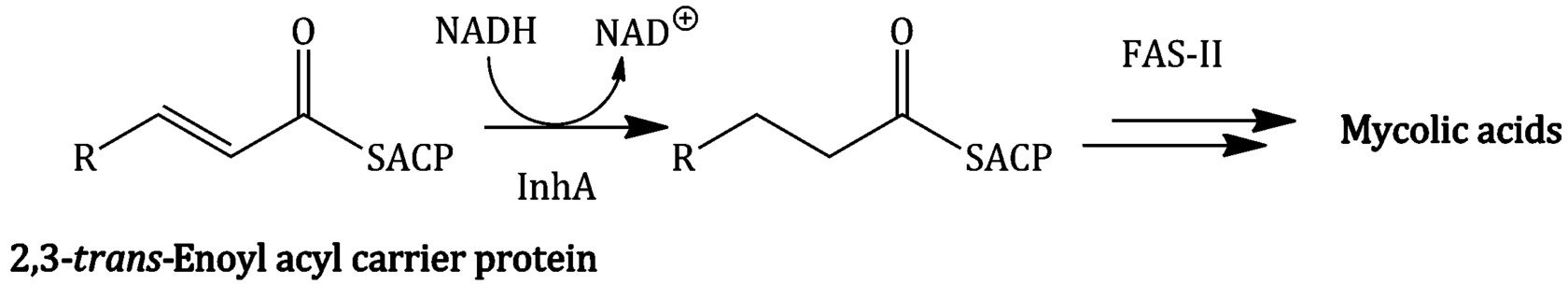
Figure 5.4.3 INH-NAD adduct bound to the active site of InhA (PDB code: 2NV6) (Vilchèze et al., 2006)
Isoniazid thus indirectly blocks the formation of the mycobacterial cell wall, through the formation of an adduct with NAD(P), which inhibits enoyl ACP reductase, thus accounting for its bacteriostatic/bactericidal activity (Kaur et al., 2009). Other mechanisms of action are still being investigated for INH, including the generation of nitric oxide (NO) radicals from INH and these radicals’ enhancement of the antimycobacterial effect of isoniazid (Timmins et al., 2004).
5.4.5 Bacterial Resistance
As might be expected, M. tuberculosis resistance to isoniazid may be the result of a number of mechanisms, including:
- Mutations/deletions in the katG gene regulatory region, resulting in a decrease in INH-NAD(P) adduct formation. This effect is not solely due to a loss of catalase-peroxidase activity, and in fact the major mutation found in clinical isolates (serine 315 to threonine) (González et al., 1999) leads to a conformational change in the substrate access channel of the enzyme, resulting in decreased isoniazid binding (Cade et al., 2010).
- Over-expression of the InhA gene, so that there is insufficient INH-NADP complex for inhibition, results in resistance in M. tuberculosis strains harbouring InhA plasmids (Larsen et al., 2002), while 25% of the mutations in the InhA gene found in clinical isolates are in the promoter or structural regions (with consequences for NADH and INH-NADP binding) (Telenti et al., 1997).
- Increased arylamine N-acetyltransferase (NAT) activity, as a result of increased expression of the NAT gene (N-acetylation reduces the activity of INH) (Payton et al., 1999).
- For more on the bacterial resistance of isoniazid, see Dye and Espinal (2001).
5.4.6 Clinical Applications
5.4.6.1 Spectrum of activity
As isoniazid targets the formation of the mycobacterial cell wall, it will not be a surprise to learn that it has activity against several types of mycobacteria – most notably against M. tuberculosis, making it a key component in the chemotherapeutic management of TB (Zhang et al., 1996). Isoniazid also shows activity against other members of the M. tuberculosis complex (MTC),19 but it lacks activity against Mycobacterium leprae (one of the causative agents of leprosy) and the Mycobacterium avium complex (MAC), and consequently should not be used to treat diseases caused by these pathogens (Inderlied et al., 1993; Eiglmeier et al., 1997). Interestingly, it is thought that the katG gene (the gene which codes for the catalase-peroxidase that activates isoniazid) of M. leprae has been inactivated, which helps explain the apparent lack of activity of isoniazid against M. leprae (and thus leprosy) (Eiglmeier et al., 1997).
5.4.6.2 Treatment of TB
Isoniazid, as discussed in Section 2.2.6.1, is used in combination with other antibacterial agents to treat active TB. Indeed, because of its high activity towards M. tuberculosis and its relatively cheap cost, it is recommended that isoniazid be used in the initial phase (alongside rifampicin, pyrazinamide, and ethambutol) and the continuation phase (alongside rifampicin) of TB treatment, unless there is a specific contraindication or bacterial resistance is suspected. Isoniazid should never be used as a monotherapy to treat active TB (although see Subsection 5.4.6.3), as this would encourage the development of resistance, which is already becoming a significant clinical problem.
TB (or consumption, as it was known – because patients were apparently consumed from within) is mostly caused by M. tuberculosis infection of pulmonary alveoli and has been responsible for many millions of deaths. The symptoms of TB are a chronic cough (with blood-tinged sputum), fever, and weight loss. If untreated, TB kills more than half of those affected. The earliest treatments for TB involved ‘fresh country air’, so many patients were sent to TB sanatoria. The dreaded ‘consumption’ has been responsible for the deaths of sufferers from every social class (including many notable figures from history, e.g. King Louis XIII of France in 1643, King Edward VI of England in 1553, General Simón Bolívar (the liberator of much of South America) in 1830, Erwin Schrödinger (the 1933 Nobel Laureate for Physics) in 1961, at least three members of the Brontë family of writers, Vivien Leigh (Scarlett O’Hara in Gone with the Wind) in 1967, and Frédéric Chopin (composer) in 1849, with countless others having contracted TB but survived (including Bishop Desmond Tutu (after whom a TB centre in South Africa has been named) and Tom Jones (whose first single was titled ‘Chills and Fever’)). Today it is estimated that one-third of the world’s population has been exposed to TB (with 90% of the exposed population having latent (asymptomatic) TB) and that there is almost one new infection per second (8.8 million new cases in 2010), with 1.4 million deaths in 2010, the majority of which were in Africa (World Health Organization, 2011). As mentioned above, resistance is a growing problem; in 2000, 3% of TB strains were multidrug resistant (MDR-TB), while in a study performed between 2000 and 2004, 20% of TB isolates were MDR and 10% were XDR (extensively drug resistant) (Centers for Disease Control and Prevention, 2006). In 2010, an estimated 650,000 cases were MDR-TB (World Health Organisation, 2011).
5.4.6.3 Prevention of TB
Isoniazid can also be used as a preventative therapy in susceptible patients who have had close contact with active TB, or those who have become tuberculin positive.20 In contrast to its use in the treatment of active TB, isoniazid can be used as monotherapy to prevent the development of active TB, and is usually taken once daily for six months.
A Cochrane review concluded that isoniazid is effective at preventing active TB in diverse at-risk patients, showing that isoniazid prophylaxis prevents the development of active TB in approximately 60% of individuals in various at-risk groups. In addition, among contacts with a positive tuberculin skin test, for every 35 patients treated with isoniazid for six months, one case of active TB will be prevented (Smieja et al., 1999). As with all drug treatments, the benefits versus risks should always be considered, however, as around 1 in every 200 patients given isoniazid prophylaxis will also develop isoniazid-induced hepatitis (Smieja et al., 1999).
One group of patients who are at increased risk of contracting TB are those infected with HIV/AIDS. Latent TB infections occur when a patient is infected with M. tuberculosis but no clinical symptoms are observed, and as we said in Subsection 5.4.6.2, it has been estimated that one-third of the world’s population has been exposed to TB and that 90% of those exposed have latent TB. In healthy individuals with a strong immune system, the latent infection is often controlled and the lifetime risk of developing active TB is low, at around 10%. However, in cases when the immune system is weakened, such as in HIV/AIDS patients, the risk of developing active disease from latent infection increases to around 30% (De Cock, 1995). Thankfully, if isoniazid is given to patients co-infected with latent TB (especially with a positive tuberculin skin test) and HIV, the risk of developing active TB is reduced (Akolo et al., 2010). More research is needed to determine the optimal duration of preventative therapy but, given the large numbers of patients co-infected with latent TB and HIV, the prophylactic use of isoniazid may have a significant impact on HIV-associated morbidity and mortality.
5.4.7 Adverse Drug Reactions
A common adverse effect associated with isoniazid is hepatic toxicity, which is associated with approximately 20% of patients receiving the drug for TB, with severe hepatotoxicity being shown in 1% of exposed patients (Scharer and Smith, 1969; Garibaldi et al., 1972).
Another serious adverse effect associated with the use of isoniazid is systemic lupus erythematosus, which occurs in approximately 1% of patients (Rubin, 2005).
As we saw in Subsection 5.4.3, isoniazid is primarily metabolised into acetylisoniazid (acetylated by N-acetyltransferase, NAT2). Individual subjects are either fast or slow acetylators (this is genetically determined); with chronic use, slow acetylation may lead to higher blood levels with increased toxicity. Acetylisoniazid is then further hydrolysed to isonicotinic acid and acetylhydrazine, which can be renally excreted. Isonicotinic acid is conjugated with glycine. Acetylhydrazine may be further metabolised to diacetylhydrazine and it is then suggested that it is converted by CYP 2E1 into hydrazine, leading to hepatotoxicity (Yard and McKennis, 1955; Ellard and Gammon, 1976; Sarich et al., 1996). Other side effects associated with isoniazid are skin rash, sideroblastic anaemia, and peripheral neuropathy, which is exhibited by those patients who are slow acetylators and/or when high doses of isoniazid are used (7.5–9.5 mg/kg) daily (Devadetta et al., 1960). The degree of neuropathy also depends on the overall nutritional state of the patient and, as a result, poorly nourished patients lacking in pyridoxine (vitamin B6, an important component in protein metabolism) are often at a higher risk (Rudman and Williams, 1983). Simple supplementation of pyridoxine in large amounts may contribute to peripheral neuropathy (Schaumberg et al., 1983), so lower doses of pyridoxine (100–200 mg daily) should be used in patients with isoniazid-induced peripheral neuropathy (Nisar et al., 1990).
Sideroblastic anaemia, in which the body has plentiful iron reserves but cannot incorporate them into haemoglobin, has been reported to have been caused by the long-term use of isoniazid (Sharp et al., 1990; Yoshimoto et al., 1992). The reasons for the poor haem incorporation into erythrocytes in sideroblastic anaemia remains elusive but isoniazid does have an effect upon delta-aminolevulinic acid synthase (ALAS), which acts upon proporphyrinogen II and subsequent haem formation (Bottomley and Muller-Eberhard, 1988). Isoniazid-induced sideroblastic anaemia is treated by withdrawing the drug and administering pyridoxine (the precursor of pyridoxal phosphate, which is a co-factor associated with ALAS function).
5.4.8 Drug Interactions
Isoniazid is metabolised in two phases (described above), in which it acts as both an inhibitor and then an inducer of the isoenzyme CYP 2E1 (Self et al., 1999). In addition, isoniazid is a weak monoamine oxidase inhibitor and has the potential to inhibit histaminase, thus causing many drug–food interactions (Hauser and Baier, 1982).
5.4.8.1 Phenytoin
There are several reports of isoniazid interacting with phenytoin, resulting in an increased serum concentration of phenytoin and, in some cases, patient toxicity (Miller et al., 1979). Isoniazid is seldom used alone and is often combined with rifampicin, which is known to induce the metabolism of phenytoin, and as such the combination of rifampicin with isoniazid in a typical TB treatment regimen will reduce the plasma levels of phenytoin (the induction effect of rifampicin upon the metabolism of phenytoin far outweighs the inhibition by isoniazid), so all patients receiving this combination of drugs need to be carefully monitored (Kay et al., 1985).
5.4.8.2 Carbamazepine
In a similar manner, carbamazepine toxicity may be experienced if given concurrently with isoniazid (Block, 1982; Fleenor et al., 1991).
5.4.8.3 Paracetamol (acetaminophen)
Isoniazid and paracetamol are both suggested to be metabolised via CYP 2E1. Isoniazid inhibits CYP 2E1 and so raises the levels of N-acetyl-para-benzoquinoneimine, which is the toxic metabolite of paracetamol, thus explaining the reports of toxicity experienced when both drugs are administered to some patients (Moulding et al., 1991; Crippin, 1993; Chien et al., 1997). The effect of slow and fast acetylators has also been investigated. Slow acetylators of isoniazid were given both paracetamol and isoniazid, and reduced levels of urinary oxidative metabolites were collected, whereas increased levels were found with rapid acetylators of isoniazid (Chien et al., 1997).
5.4.8.4 Benzodiazepines
Isoniazid treatment causes hepatic impairment of the N-demethylation of diazepam, resulting in a prolonged half-life of this benzodiazepine (Ochs et al., 1981). Isoniazid also increased the half-life of triazolam, but not oxazepam, as the latter is metabolised by glucuronidation only, thus avoiding the interaction with isoniazid (Ochs et al., 1983).
5.4.8.5 Vitamin D
Vitamin D is hydroxylated, in both the liver and the kidneys, by CYP 27B1, and a study has shown that the administration of isoniazid reduced the serum concentration of calcium, 25-hydroxyvitamin D, and 1α,25-dihydroxyvitamin D (Brodie et al., 1981).
5.4.8.6 Foods
As mentioned previously, isoniazid is a known monoamine oxidase inhibitor and so results in the reduced clearance/metabolism of foods high in monoamines, such as tyramine (Hauser and Baier, 1982). High plasma levels of tyramine (found in strong cheeses, etc.) may cause flushing, palpitations, and headache, with sometimes an increase in systolic blood pressure, so these foods should be avoided in patients taking isoniazid.
As isoniazid is also a histaminase inhibitor, foods high in histamine (e.g. tuna, shellfish, cured pork, parmesan cheese) should also be avoided, due to the reduced metabolism of histamine (Baciewicz and Self, 1985).
5.4.9 Recent Developments
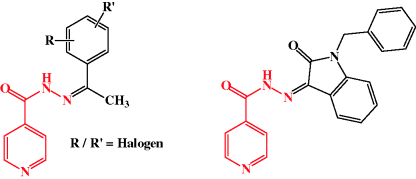
Surprisingly, given the advent of MDR-TB (when TB is resistant to isoniazid and rifampicin), research into developing isoniazid derivatives is not a hot area at present. Many pharmaceutical companies are focussing on developing novel agents for the treatment of TB rather than expanding an existing class – especially one that has been around since the 1950s. There have been several attempts to prepare isoniazid derivatives, with one group preparing halogenated analogues (Vigorita et al., 1994) and, more recently, another group preparing a set of isatin (indolin-2,3-dione) derivatives (Figure 5.4.4) (Aboul-Fadl et al., 2003). When tested in vitro against M. tuberculosis, the halogenated analogues had inferior activity to isoniazid, but several isatin derivatives showed activity against an isoniazid-resistant strain of M. tuberculosis. Further work has shown that the isatin derivatives only have minor effects on mouse hepatic enzymatic activity and are thus likely to be free of major CYP drug–drug interactions (Franklin et al., 2010), although it remains to be seen what role these compounds might have in the actual management of TB.
Figure 5.4.4 Halogenated isoniazid analogues (left) and an example of an isatin-isoniazid derivative (left). The structure of the isoniazid scaffold is shown in red
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


