The field of iron biology has made rapid advances since 2000. Many of the proteins involved in iron transport and homeostatic regulation have been identified, and their physiologic roles have been uncovered. Perhaps the most exciting breakthrough came with the discovery of iron regulatory hormone, hepcidin. Features of hepcidin regulation of iron metabolism have been compared to insulin action in glucose metabolism, thus creating a new field of iron endocrinology (
10). How hepcidin regulates systemic iron homeostasis is a major focus of current investigation. At the cellular level, molecular insights into the regulation of iron-binding proteins have yielded information about regulation of iron transport, utilization, and storage that have important clinical considerations. Finally, transcriptional and posttranscriptional networks that may be activated by hepcidin-induced signaling are beginning to emerge and provide clues into the relationships between iron and inflammation.
Intestinal Iron Absorption
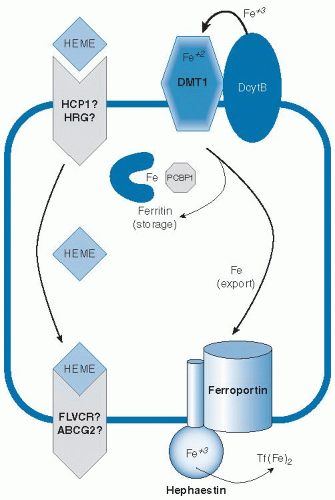
Because body iron status is precisely tuned by the absorption of dietary iron, it is important to understand the mechanisms involved in this process and the multiple layers that regulate the flux of iron into the system (
Fig. 10.2). Because the body does not eliminate excess iron, dysregulation of intestinal iron absorption causing assimilation of too much iron will result in iron overload. Conversely, if sufficient iron is not absorbed to make up for small daily losses, the risk for iron deficiency increases. Iron is absorbed as nonheme or heme forms. Heme iron is absorbed more effectively (
11), and this process does not appear to be subject to the same regulatory mechanisms as nonheme iron uptake (
12).
These findings indicate that heme and nonheme iron are taken up by independent mechanisms. A putative heme transporter called heme carrier protein-1 (HCP1)
was identified (
13), but questions about its true function arose when a role in folate transport was determined for the same factor (
14). HCP1 may possibly be a low-affinity transporter for heme, but its physiologic relevance remains to be better established. A different molecule, heme-responsive gene-1 or
HRG1, has been identified as a heme transporter in
Caenorhabditis elegans (
15). Although a similar gene is present in humans, its activity has yet to be defined. Heme oxygenase may release iron from heme entering the intestinal absorptive enterocyte to join the pool of newly absorbed nonheme iron entering the cell (
16). Alternatively, intact heme may be released across the basolateral surface. Feline leukemia virus C receptor (FLVCR) has been identified to function as a heme exporter in erythroid cells (
17), and a second possible efflux pathway involving the ABC transporter ABCG2 (also known as breast cancer regulated protein, BCRP) has been suggested (
18), but their possible roles in heme assimilation by the intestine have yet to be fully explored.
Uptake of nonheme iron by enterocytes is better understood. Although less effectively absorbed, nonheme iron is present in a greater range of foods, most typically in the ferric (Fe
3+) form. Reduction to Fe
2+ is the first step in intestinal nonheme iron assimilation and is mediated by brush-border ferrireductase activity. An enzyme called duodenal cytochrome B (DcytB) has been implicated in this process (
19). Although it does not appear to be an essential gene (
20), DcytB is highly regulated in response to iron status in animals and humans (
21,
22,
23), and a promoter polymorphism observed in the human population appears to modify serum ferritin levels in
HFE-associated hereditary hemochromatosis (
24). After reduction by DctyB or another brush-border ferrireductase, uptake of the ferrous (Fe
2+) form of iron is mediated by divalent metal transporter-1 (DMT1) (
25,
26,
27). The low pH of the intestinal lumen is important for these initial steps because DMT1 is a protoncoupled transporter, and acidification is therefore necessary for its optimal activity (
26). Like DcytB, DMT1 is also highly regulated by iron status. The small intestine expresses four transcripts of DMT1, and mRNA levels are regulated both transcriptionally and posttranscriptionally (
28,
29).
Different isoforms of the protein appear to have tissuespecific function and subcellular localization (
30). Iron status appears to control not only the protein and mRNA levels for DMT1 but also the distribution of the protein in various enterocyte compartments (
31). Studies have shown that intestinal DMT1 is necessary for iron absorption in mice after birth, but it appears to be dispensable for other tissues—a finding suggesting that redundant activities fulfill that role (
27). Human mutations in DMT1 are associated with microcytic anemia (
32), consistent with its major function in dietary iron absorption. The finding that these patients also load iron is consistent with an important DMT1 function in delivering iron to erythroid cells (see the later description of the transferrin cycle).
Molecular details of the transfer of imported nonheme iron across the intestinal mucosal cell for release into circulation have also emerged. Investigators have long speculated that a cytosolic iron chaperone directs the fate of newly absorbed intestinal iron. Only one such factor has been identified to date, however, and its function in the intestine has yet to be fully characterized. Poly(rC)-binding protein-1 (PCBP1) has been shown to deliver iron to ferritin and is ubiquitously expressed (
33). When efflux from the enterocyte is impaired, intestinal iron is known to accumulate in the ferritin storage compartment (
34,
35). Deletion of intestinal ferritin in mice promotes increased dietary iron absorption and dysregulated systemic iron metabolism (
36). Ferritin iron stored as a result of excess dietary absorption would probably be lost from the body as enterocytes are shed from the villus tip. It seems likely that PCBP1 may function in the intestine to help load iron onto ferritin. Whether an iron chaperone function is necessary to traffic cytosolic iron across the mucosa for its entry into portal circulation is unclear, however.
An alternate model is that iron crosses the enterocyte through a vesicular trafficking pathway (
37,
38), which involves intracellular transfer to a compartment with the membrane iron exporter ferroportin and the ferroxidase hephaestin, along the ultimate target iron-free apotransferrin. Each of these factors plays an important role in export of iron from the enterocyte, but whether these factors act within the lumen of intracellular vesicles or directly at the basolateral surface is not certain because they are topologically equivalent. Ferroportin is essential for iron efflux from the intestine (
39). It is believed to export iron in concert with hephaestin, a membranebound ceruloplasmin homolog that oxidizes ferrous iron to the ferric form (
35). Ceruloplasmin itself can also fulfill this function (
40), and both ferroxidases provide iron to transferrin in the correct oxidation state. Transferrin binds two atoms of ferric iron and circulates in serum to deliver iron to peripheral tissues. In fact, fasting transferrin saturation is recommended as the most sensitive serum index of iron status because postprandial increases can otherwise cause false-positive indications of iron-loading (
41).
The Transferrin Cycle
Circulating transferrin delivers iron by binding to cell surface receptors. Two receptors that specifically and uniquely recognize transferrin as a ligand are known. Transferrin receptor-1 has long been studied as the functional partner in iron uptake and is ubiquitously expressed (
42,
43). Its closely related homolog, transferrin receptor-2, has a more restricted expression pattern and predominates in liver, where it plays an iron-sensing role in metabolism (
44,
45,
46).
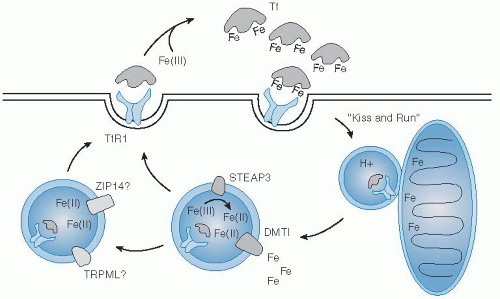
Uptake of iron by cells from the transferrin-receptor binding complex begins with its internalization by clathrin-mediated endocytosis (
Fig. 10.3). Clathrin-coated vesicles deliver their cargo to acidic intracellular compartments called early endosomes. The low pH of this environment promotes release of iron and stabilizes the binding of apotransferrin
to the receptor. Together, they are recycled back to the cell surface, where apotransferrin dissociates from the receptor at neutral pH (
47). Reduction of ferric iron released in the lumen of the endosome is supported by the ferrireductase Steap3 (
48). Subsequent transport of ferrous iron in erythroid cells is mediated by DMT1 (
49). In one model, investigators proposed that reticulocyte endosomes bearing iron-bound transferrin directly deliver cargo to mitochondria for heme biosynthesis, the “kiss-and-run hypothesis” (
50).
Still, other transporters may provide for iron transfer in the endosomal-lysosomal system in peripheral tissues, including Zip14 (
51) and TRPML1 (
52). On entering the cell cytoplasm, iron is either rapidly metabolized or stored in ferritin. A small labile iron pool exists and is in the micromolar range in most cell types. In excess, this free iron can produce reactive oxygen species that can cause cellular damage. For example, loss of iron storage as a result of tissue-specific deletion of ferritin heavy chain in mice leads to liver damage (
53). Defects in transferrin’s intracellular trafficking are also known to give rise to anemia in the
hbd hemoglobin-deficit mouse model as a result of mutation of Sec15l1 of the exocyst complex (
54). Emerging evidence indicates that the iron-regulated myotonic dystrophy kinase-related CDC42-binding kinase α (MRCKα) may be involved in modulating transferrin-mediated iron uptake, possibly by its association with the actin cytoskeletal network and the transferrin-transferrin receptor complex (
55). Clearly, the transferrin cycle must be regulated tightly and in a coordinated manner to control the distribution of iron sufficient to meet metabolic demands but limited to avoid toxicity.