INTRODUCTION
LEARNING OBJECTIVES
After studying this chapter you should be able to:
Construct a flow diagram depicting iron homeostasis: absorption from the gut, transport in the plasma, incorporation into erythroid precursors, release from senescent red cells, and return to the plasma.
Explain the important role of hepcidin in iron absorption from the gut and release of iron from macrophages.
Name and prioritize the causes of iron deficiency.
Describe the key laboratory features of iron deficiency.
Identify the causes of iron overload and its clinical manifestations.
In all organisms from bacteria to man, iron is by far the most important metallic element. Its outer shell of electrons is ideally poised for complex coordination chemistry, enabling the binding of ligands such as oxygen as well as participation in critical oxidation-reduction reactions. Iron is essential not only for the biological activity of heme proteins such as hemoglobin, myoglobin, and cytochromes but also as a key cofactor in a number of enzymes spanning a wide range of metabolism. However, because of iron’s high degree of reactivity, it can catalyze the generation of oxygen free radicals and other toxic species, leading to cellular and tissue injury by way of protein cross-links, lipid peroxidation, and damage to DNA. Therefore, in order for iron to safely fulfill its biological functions, an exquisite degree of control is required. In this chapter we will review the basic elements of iron homeostasis: absorption, transport, utilization, recycling, and excretion. During the last decade, understanding of these processes has been enormously enhanced by the molecular cloning and characterization of critical genes, some of which were discovered by investigation of mutant mice and zebra fish whose phenotypes suggested perturbed iron metabolism.
This chapter will begin with an overview of iron homeostasis, as this information is essential for understanding the pathogenesis, clinical features, and treatment of iron deficiency and iron overload.
NORMAL IRON HOMEOSTASIS
Safe and effective transport and utilization of iron are achieved by tight regulation at the level of both individual cells and the organism as a whole. The expression of a number of proteins that play critical roles in iron metabolism is regulated by the intracellular concentration of iron. This is achieved through a consensus stem loop sequence in the messenger ribonucleic acids (mRNAs) that encode these proteins. When iron is scarce, two iron regulatory proteins (IRPs) bind specifically to this stem loop and modify either the stability or rate of translation of the mRNAs, whereas when intracellular iron is abundant, the IRPs assume a conformation that precludes mRNA binding. Systemic iron metabolism is regulated by the circulating polypeptide hormone hepcidin, which controls both dietary iron absorption from the gut and release of recycled iron from macrophages. These two modes of regulation are described in detail later in this chapter.
The dietary sources of iron vary considerably according to geographic location, cultural tastes, and economic status. Iron in food consists of inorganic salts and organic complexes derived from plants as well as heme from animal sources. Digestion of grains, vegetables, and fruits in the stomach and duodenum results in the release of ferric iron. As depicted in Figure 5-1, normal individuals absorb only 1 to 2 mg of iron per day, primarily at the villous tips of duodenal enterocytes. A ferrireductase at this site reduces the iron to its ferrous form, allowing it to enter the cell through a luminal transmembrane channel, the divalent metal transporter DMT1 (Fig. 5-2). A portion of the iron that has entered the enterocyte may be stored within a porous multimeric protein cage called ferritin. Ferrous iron exits from the cell through the transport protein ferroportin, which is localized within the plasma membrane on the abluminal or basolateral side of the enterocyte. Here the iron is rapidly oxidized to the ferric form that binds transferrin, the plasma protein responsible for iron transport throughout the circulation. As shown in Figure 5-2, the export of iron from the duodenal enterocyte can be suppressed by hepcidin, a small polypeptide hormone produced in the liver. The binding of hepcidin to its receptor, ferroportin, triggers the latter’s internalization and subsequent degradation. As a result, the rate of egress of iron from the enterocyte is markedly dampened.
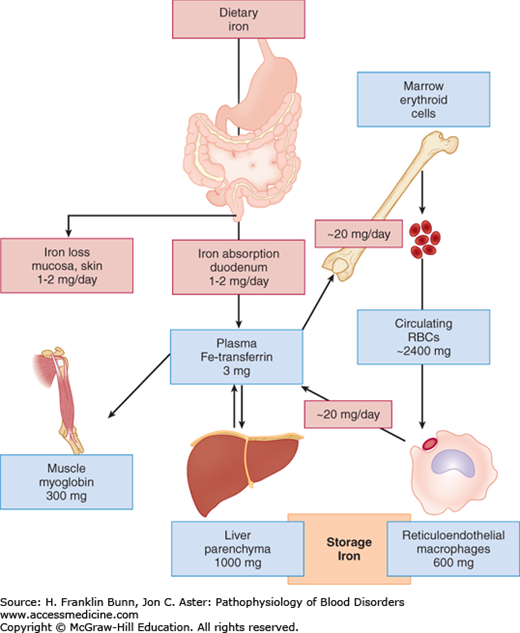
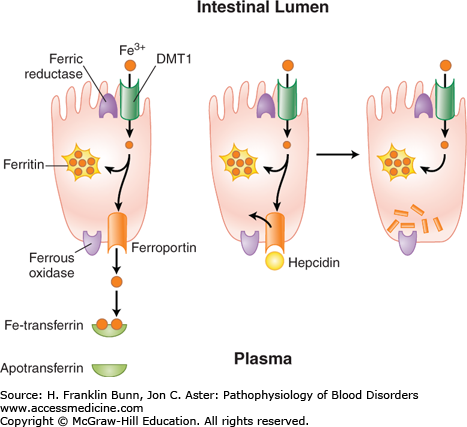
FIGURE 5-2
Absorption and egress of iron in the duodenal enterocyte. Iron (Fe3+) within the duodenal lumen is reduced to Fe2+ which enters the cell through divalent metal transporter (DMT1) and exits the cell on the other (abluminal) side through ferroportin and is oxidized to Fe3+. The binding of the master regulator hepcidin to ferroportin (middle) causes rapid internalization and degradation (right) of ferroportin, thereby blocking further egress of iron from the cell.
Among individuals from North and South America as well as Europe, a substantial portion of dietary iron is in the form of hemoglobin and myoglobin within meat and other animal sources. Following proteolytic degradation of these proteins in the stomach and proximal intestine, heme is released and absorbed intact into the enterocyte, where it is degraded by heme oxygenase, resulting in the release of iron that is either stored as ferritin or exits from the cell via ferroportin. The luminal heme channel/importer has not yet been identified.
The plasma protein transferrin binds ferric iron at two sites with extraordinarily high affinity. As a result of such tight binding, the concentration of free iron in the plasma is too low to be easily measured. Thus transferrin effectively protects tissues and cells from the toxicity of “free” iron. As shown in Figure 5-1, transferrin picks up iron either from duodenal enterocytes or from macrophages, which (as discussed later in the “Iron Recycling” section) accumulate iron recycled from hemoglobin in senescent red cells.
All cells except the duodenal enterocyte take up iron via the binding of Fe-transferrin to transferrin receptors on the plasma membrane. In keeping with their obligation to produce high levels of hemoglobin, erythroid precursor cells express much higher levels of transferrin receptors than do other cells. As illustrated in Figure 5-3, the expression of transferrin receptor is regulated in an iron-dependent manner by the binding of IRPs to earlier-mentioned consensus stem loops at the 3′ untranslated region of transferrin receptor mRNA. In iron deficiency, when there is a premium on optimizing entry of iron into cells, the binding of IRPs to these stem loops greatly enhances the stability of the transferrin receptor mRNA and thereby increases protein expression. Up-regulation of the transport protein DMT1 in iron deficiency also depends on enhanced mRNA stability via binding of IRPs to 3′ mRNA stem loops.
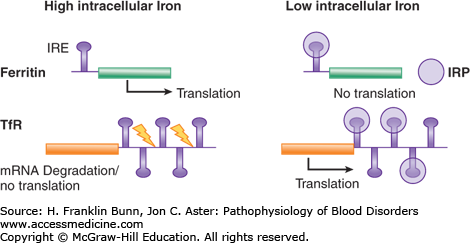
FIGURE 5-3
Iron-dependent regulation of the rate of translation of ferritin messenger ribonucleic acid (mRNA) and the stability of transferrin receptor (TfR) mRNA. When the intracellular iron level is low, iron regulatory proteins (IRPs) bind to a consensus stem loop iron regulatory element in the respective mRNAs, resulting in enhancement of transferrin receptor mRNA levels and suppression of ferritin translation. When the intracellular iron level is high, the IRPs do not bind to the stem loops. Transferrin receptor mRNA is degraded (yellow arrows), and translation of ferritin is unimpeded.
Transferrin molecules loaded with two iron atoms have much greater affinity for the receptor than do those with either a single atom or none. As shown in Figure 5-4, the iron-transferrin/transferrin receptor complex is rapidly internalized as a plasma membrane microvesicle that includes the iron transport protein DMT1. A proton pump docks onto this microvesicle and acidifies its interior, thereby releasing iron, which then exits via DMT1. Once in the cytosol, this iron either goes to mitochondria for heme biosynthesis or is stored as ferritin.
FIGURE 5-4
The uptake of transferrin-bound iron (Fe2-Tf) into a cell via the transferrin receptor (TfR). Microvesicles containing the Fe2
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


