Chapter 1
Introduction
1.1 What is biochemical pharmacology?
What is it?
- pharmacology, but with a focus on how drugs work, not on whether we should take them before or after dinner
- fascinating—you will love it, or double your money back
What is it not?
- just molecular pharmacology—physiological context is important, too
- a claim that we completely understand the biochemical action modes of all practically useful drugs—we don’t
Notes: Let me repeat the last two points—the action modes of many drugs are incompletely understood, and physiology is important. I will try to cover the key aspects of physiology as far as they are needed to understand the course material, but you may find it useful to consult an actual physiology text on the side from time to time.
1.1.1 On drugs and poisons: Paracelsus’ maxim
Notes: In his day, Paracelsus acquired a reputation not just as a physician, but also for his fondness of a good drink; rumor has it that this latter inclination was useful to him in his discovery of the above maxim.
As you know, many drugs turn poisonous if overdosed, and many poisonous natural compounds may become useful as drugs when properly diluted. That holds for both legal and illegal drugs; we will consider examples of both classes in this course.
“Alle Ding’ sind Gift und nichts ohn’ Gift; allein die Dosis macht, dass ein Ding kein Gift ist.”
“All things are poison and nothing is without poison; only the dosage makes it so that something is not a poison.”
“Dosis sola facit velenum.”

1.1.2 A very small drug particle and a very large one
Notes: Lithium is the smallest drug particle—it is not even a molecule but just a single atom. It is also an example of a drug whose mode of action is still unknown, despite many decades of research; various biochemical effects have been described, but none of these has been conclusively linked to its antidepressant activity.
Urokinase is a protein and as such is much larger than most other drug molecules. It is used to dissolve intravascular blood clots in order to restore perfusion in acute myocardial infarction and stroke.
Note that neither lithium and urokinase are very typical drugs; some more typical ones are shown in the next slide.
1.1.3 Some drug molecules of more typical size
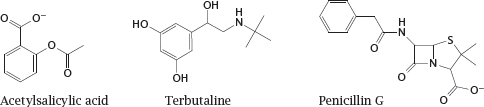
Notes: Most drugs are organic molecules with molecular weights of no more than 500 Da, such as the examples shown here. This rule of thumb holds for drugs of very different functional classes.1 As you may know, acetylsalicylic acid inhibits cyclooxygenase, and penicillin inhibits bacterial cell wall synthesis; terbutaline activates β2–adrenergic receptors. We will discuss all these drug actions in more detail in later chapters.
Organic drug molecules may be natural compounds or obtained through organic synthesis. Some drugs are obtained semisynthetically, that is, through the synthetic modification of natural compounds; for example, penicillin G and other penicillin derivatives are obtained through semisynthesis.
1.2 Drugs and drug targets
Practically all drugs—with the exception of those that act osmotically; see below—must bind to target molecules in order to exercise their effects. Most drugs target proteins.
1.2.1 Functional classes of protein drug targets
Notes: As we work through the example drugs in this course, you will notice that most of the drug targets fall into one of the above categories.
1.2.2 Non-protein drug targets
Notes: Drug targets other than proteins are less common. DNA is important with anticancer drugs. The RNA of bacterial ribosomes is targeted by various classes of antibiotics. It is noteworthy, however, that many regulatory effects of RNA have only recently been discovered and appreciated. In experimental research, it has become quite common to inhibit the expression of specific genes at the level of mRNA, and in a few cases this strategy has already found clinical application. This is discussed in more detail in chapter 13.
Lipid membranes are targeted again by some antibiotics, which recognize specific lipids that occur in microbial cell membranes but not mammalian ones. The accumulation of alcohol and of gaseous narcotics in the cell membranes of neurons may contribute to the effects of these compounds; the long-standing question as to what extent these effects arise from direct interaction with membrane proteins as opposed to membrane lipids has not yet been fully resolved.
Osmotically active drugs are used in order to influence the distribution of fluids between compartments. An important application is the use of dextran or hydroxyethylstarch as blood plasma expanders, that is, to replace lost blood volume. The osmotic activity of such macromolecules counteracts the hydrostatic pressure difference that exists across the capillary walls of the circulation. If lost blood were replaced with physiological saline solution instead, this fluid would rapidly leak out into the tissues due to the intracapillary pressure.
While plasma expanders are clinically important, they are not very interesting from a biochemical point of view, and therefore we will not consider them any further.
1.2.3 Histamine receptor antagonists
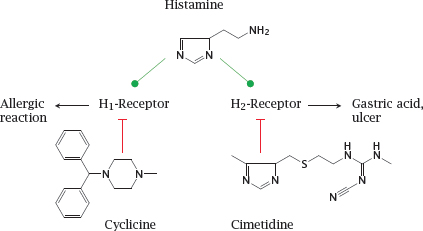
Notes: Among the hormone and neurotransmitter receptors, a particularly prominent class are the G protein-coupled receptors (GPCRs). These receptors are found in the cytoplasmic membranes of all kinds of cells and respond to a large number of different ligands. It is often said that approximately 50% of all clinically used drugs target G protein-coupled receptors. We will devote all of Chapter 5 to this important receptor class; here, we will briefly look at histamine receptors and angiotensin receptors as examples.
Histamine receptors occur in several subtypes. While all subtypes are activated by the same physiological ligand—that is, histamine—they may differ in their susceptibility to inhibition by receptor antagonists. For example, cyclizine inhibits H1-receptors, whereas cimetidine selectively blocks H2-receptors. Subtype-selective drugs are often useful in pharmacotherapy; for example, H1-selective inhibitors can be used to treat allergies without interfering with acid secretion, and H2-selective drugs can be used to treat gastric hyperacidity without causing drowsiness, which is a common side effect of H1-receptor blockade.
While cyclizine has only remote structural similarity to histamine, the resemblance is more obvious with cimetidine. This similarity is not coincidental.
1.2.4 The development of H2-receptor blockers
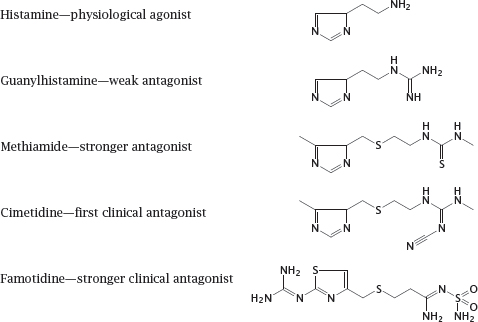
Notes: The development of cimetidine began in the 1960s. The histamine receptors had not been molecularly characterized at the time; all that was known about them was that they existed and were activated by histamine, and that some of them were involved in the secretion of gastric acid. Gastric acid was known to be a factor in the causation of gastric ulcers.2 The guiding idea was to modify the structure of histamine so as to turn it from an activator into an inhibitor, which could then be used to treat the ulcers.
This plan proved extraordinarily successful. The slide illustrates intermediate stages of the development process. Guanylhistamine was the first derivative that retained affinity for the receptor but inhibited it. Further modification yielded cimetidine, which improved upon the receptor affinity of guanylhistamine and became the first clinically used H2-receptor antagonist, serving as the mainstay of ulcer therapy for more than a decade. Even more avidly binding antagonists, such as famotidine, can be administered at dosages of just a few milligrams per day (as compared to several hundreds of milligrams of cimetidine).
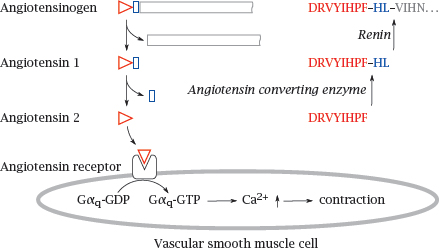
1.2.5 Angiotensin: Proteolytic release from angiotensinogen, and mode of action