1Abbreviations: ABC, adenosine triphosphate-binding cassette; ACAD, acyl-CoA dehydrogenase; ACD, acyl-coenzyme A dehydrogenase; AFLP, acute fatty liver of pregnancy; ATP, adenosine triphosphate; CACT, carnitine-acylcarnitine translocase; CoA, coenzyme A; CPT, carnitine palmitoyltransferase; DHA, docosahexaenoic acid; EFA, essential fatty acid; ETF, electron transfer flavoprotein; FAD, flavin adenine dinucleotide; GA, glutaric aciduria; HELLP, hemolysis, elevated liver enzymes, low platelet; HMG, hydroxymethylglutaryl; LCAD, long-chain acyl-coenzyme A dehydrogenase; LCHAD, long-chain 3-hydroxyacyl-coenzyme A dehydrogenase; MADD, multiple acyl-coenzyme A dehydrogenation disorder; MCAD, medium-chain acyl-coenzyme A dehydrogenase; MCT, medium-chain triglyceride; MRI, magnetic resonance imaging; PKU, phenylketonuria; PTS, peroxisomal targeting signal; RCDP, rhizomelic chondrodysplasia punctata; SCAD, short-chain acyl-coenzyme A dehydrogenase; SCOT, succinyl-coenzyme A:3-ketoacid coenzyme A transferase; SIDS, sudden infant death syndrome; TFP, trifunctional protein; VLCAD, very-long-chain acyl-coenzyme A dehydrogenase; VLCFA, very-long-chain fatty acid; X-ALD, X-linked adrenoleukodystrophy.
β-Oxidation, which results in sequential cleavage of two carbon units from fatty acids, represents an important source of energy for the body during times of fasting and metabolic stress. Free fatty acids released into the blood by catabolism of fat stores or from dietary sources are metabolized in the mitochondria. β-Oxidation also functions as a degradative route for complex lipids in a different subcellular compartment, the peroxisomes (sometimes referred to as microbodies). Peroxisomes are subcellular organelles bounded by a single lipid bilayer membrane (1). They are ubiquitously distributed in tissues but are particularly abundant in liver and kidney (2).
All peroxisomal proteins are encoded in the nuclear genome, synthesized on free cytoplasmic polyribosomes, and transported to the organelle posttranslationally. This process is mediated by specific targeting sequences on proteins and a variety of specific receptor proteins on the peroxisomes (3, 4, 5, 6). The most common is the presence of a serine-lysine-leucine amino acid motif at the carboxy terminus of the protein. This peroxisomal targeting signal (PTS1), present in more than 95% of proteins destined for the peroxisomal matrix, binds to the cytosolic receptor protein Pex5p. A second mechanism uses an amino terminus targeting signal (PTS2) that binds to the receptor protein Pex7p. The PTS-receptor complexes are stabilized and transported to the peroxisomal membrane, where they interact with the docking machinery and are then translocated into the peroxisomal matrix. To date, 32 proteins involved in these processes, encoded by PEX genes and known as peroxins, have been identified and characterized.
Mitochondria are bounded by two lipid bilayer membranes (the inner and outer mitochondrial membranes) (7). The intermembrane space constitutes a distinct compartment within the mitochondria, whereas the space bounded by the inner mitochondrial membrane is known as the matrix. Mitochondria are unique organelles in animals in that they contain their own genetic information and are solely maternally inherited (8). Most proteins found in the mitochondria, however, are nuclear encoded and thus are inherited in a standard Mendelian fashion. In general, they are synthesized in a larger precursor form containing information in an amino terminal signal peptide necessary for targeting the proteins to the mitochondria (9, 10). These signal sequences are usually removed after import of the protein into the mitochondrion (11). More than one targeting signal may be necessary to direct the imported protein to the correct mitochondrial space or membrane.
Peroxisomes and mitochondria arise by division of previously existing organelles and are randomly distributed to daughter cells on cellular division (12, 13). Peroxisomes interact with mitochondria at multiple levels. They share common fission factors, which are involved in the division of both peroxisomes and mitochondria. Peroxisomes are metabolically linked to mitochondria through the β-oxidation of fatty acids and the metabolism of reactive oxygen species. Secondary mitochondrial changes occur in some peroxisomal biogenesis defects (14, 15).
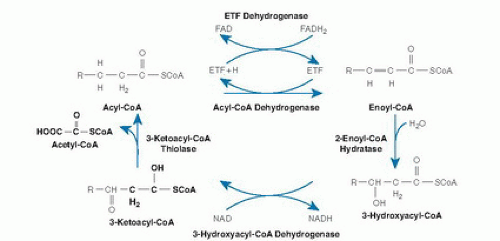
Mitochondrial β-oxidation is predominantly responsible for the oxidation of fatty acids of carbon length 20 or less (16, 17). β-Oxidation is a complex process involving transport of activated acyl-coenzyme A (CoA) moieties into the mitochondria and sequential removal of 2 carbon acetyl-CoA units (Fig. 70.1) (18). The pathway of mitochondrial fatty acid oxidation is initiated by activation of fatty acids to acyl-CoA esters in the cytosol. The fatty acids are then transferred across the mitochondrial membrane bound to carnitine. Within the mitochondrial matrix, the acylcarnitines are converted back to acyl-CoAs. The 4 steps of the β-oxidation cycle then sequentially remove 2 carbons until the acyl-CoA (n carbons) is fully converted to n/2 acetyl-CoA molecules. In peripheral tissues, the acetyl-CoA is terminally oxidized in the Krebs cycle for adenosine triphosphate (ATP) production. In the liver, the acetyl-CoA from fatty acid oxidation can instead be used for the synthesis of ketones, 3-hydroxybutyrate, and acetoacetate, which are then exported for final oxidation by brain and other tissues (Fig. 70.2) (19).
Fig. 70.1. Pathway of enzyme and transporter proteins involved in mitochondrial β-oxidation. CoA, coenzyme A; ETF, electron transfer flavoprotein; FAD, flavin adenine dinucleotide; NAD, nicotinamide adenine dinucleotide. (Modified and reprinted with permission of the Mayo Clinic and Foundation from Vockley J. The changing face of disorders of fatty acid oxidation. Mayo Clin Proc 1994;69:249-57.)
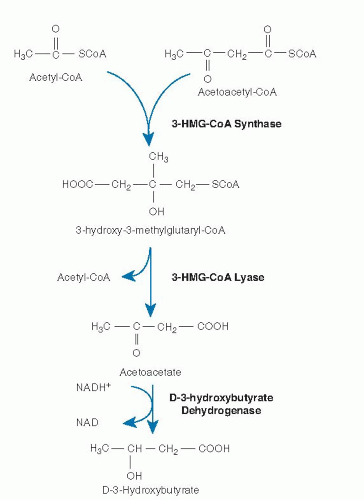
Fig. 70.2. Generation of ketone bodies from the products of β-oxidation. CoA, coenzyme A; HMG, hydroxymethylglutaryl; NAD, nicotinamide adenine dinucleotide.
At least 25 enzymes and specific transport proteins are responsible for carrying out the steps of mitochondrial fatty acid metabolism, some of which have only recently been recognized (see Fig. 70.1 and Table 70.1) (10). Of these, defects in at least 22 have been shown to cause disease in humans (10).
ENZYMES OF β-OXIDATION
Mitochondria
Free fatty acids are transported through the blood after intestinal absorption or mobilization from endogenous stores by the use of albumin as a carrier protein or in the form of triacylglycerols in lipoprotein complexes (20). Transport of free fatty acids intracellularly and through the cytoplasm is probably accomplished by a specific transport process; however, the mechanism of this step is not well characterized (21). Before undergoing β-oxidation, free fatty acids must be activated to their corresponding acyl-CoA thioesters. Long-chain specific acyl-CoA synthetases can be found in various subcellular locations but are thought to arise from a single gene product (22). Short- and medium-chain carboxylic acids directly enter the mitochondrial matrix, where they are activated. In contrast, long-chain fats are activated in the cytoplasm and require active transport into mitochondria. Transport of long-chain acyl-CoAs requires at least two enzymes, a transporter protein and the use of carnitine as an intermediate carrier molecule. Carnitine is itself transported intracellularly by a specific transporter protein (23). Two carnitine transporters have been described, one specific to liver and a second with a more ubiquitous distribution, including kidney, muscle, and fibroblasts. Longchain acyl-CoAs are conjugated to carnitine by carnitine palmitoyltransferase I (CPT I). This enzyme is located on the inner aspect of the outer mitochondrial membrane. Tissue-specific isoforms of this enzyme exist for muscle, liver, and brain (23). Long-chain acylcarnitines are then passed to carnitine palmitoyltransferase II (CPT II) in the inner mitochondrial membrane by a translocase.
TABLE 70.1 ENZYMES INVOLVED IN MITOCHONDRIAL FATTY ACID OXIDATION
ENZYME
PROVEN CLINICAL DISORDER
Fatty acid activation
Acyl-CoA synthetase
No
Carnitine cycle
Plasma membrane carnitine transporter
Yes
CPT I
Yes
Carnitine-acylcarnitine translocase
Yes
CPT II
Yes
Mitochondrial β-oxidation spiral
Very long-chain acyl-CoA dehydrogenase (membrane)
Yes
LCAD (matrix)
No
MCAD
Yes
SCAD
Yes
Trifunctional protein
Yes
Long-chain 2-enoyl CoA hydratase
Long-chain 3-hydroxyacyl-CoA dehydrogenase
Yes (isolated)
Long-chain 3-ketoacyl-CoA thiolase
Crotonase (short-chain 2-enoyl-CoA hydratase)
No
SCHAD
Yes
Short-chain 3-ketoacyl-CoA thiolase
Possible
Enzymes of β-oxidation of unsaturated fats
Long-chain Δ3,Δ2-enoyl-CoA isomerase
No
Short-chain Δ3,Δ2-enoyl-CoA isomerase
No
2,4-Dienoyl-CoA reductase
Possible
Enzymes of ketone body production
HMG-CoA synthase
Yes
HMG-CoA lyase
Yes
D-3-Hydroxybutyrate dehydrogenase
CoA, coenzyme A; CPT, carnitine palmitoyltransferase; HMG, hydroxymethylglutaryl; LCAD, long-chain acyl-coenzyme A dehydrogenase; MCAD, medium-chain acyl-coenzyme A dehydrogenase; SCHAD, short-chain 3-hydroxyacyl-coenzyme A dehydrogenase; SCAD, short-chain acyl-coenzyme A dehydrogenase;.
Once present in the mitochondrial matrix, acyl-CoAs of all chain lengths undergo a series of enzymatic reactions that results in the release of the two carbon unit acetyl- CoAs and a new acyl-CoA molecule that is two carbons shorter. The first step in this cycle is the dehydrogenation of the acyl-CoA to 2-enoyl-CoA. This reaction is catalyzed by a family of related enzymes, the acyl-CoA dehydrogenases (ACAD) (24). Four different members of this family are active in β-oxidation: very-long-, long-, medium-, and short-chain acyl-CoA dehydrogenases (very-longchain acyl-CoA dehydrogenase [VLCAD], long-chain acyl-CoA dehydrogenase [LCAD], medium-chain acyl-CoA dehydrogenase [MCAD], and short-chain acyl-CoA dehydrogenase [SCAD], respectively), which differ in their chain length specificity. The role of LCAD in fatty acid β-oxidation is unclear. LCAD is present in much lower concentrations than VLCAD in tissues where the two have been separated and thus would appear to play a minor role in the flux of fatty acids through β-oxidation. However, LCAD has significant activity toward long, branched-chain substrates, unlike VLCAD, and thus may be more important in their metabolism (25, 26). A final long-chain ACAD, designated ACAD9, is more active toward unsaturated substrates than saturated ones, but its full role in cellular metabolism is not yet clear (27).
The acyl-coenzyme A dehydrogenases (ACDs) differ from most other dehydrogenases because they use electron transfer flavoprotein (ETF) as a final electron acceptor and thus can channel electrons directly into the ubiquinone pool of the electron transport machinery by way of ETF:ubiquinone oxidoreductase (ETF dehydrogenase) (28). The ACDs are homotetramers (except VLCAD and ACAD9, which are homodimers) whose monomers are synthesized in a larger precursor form in the cytoplasm from nuclear encoded transcripts and are then transported into mitochondria (24, 29). Once inside the mitochondrial matrix, the leader peptide is removed by a specific protease; and the mature subunits assemble into the active multimer. One molecule of flavin adenine dinucleotide (FAD) is noncovalently attached to each ACD subunit. cDNAs for each of these proteins have been cloned, and sequence analysis shows that they are approximately 30% to 35% conserved, a finding suggesting evolution from a common primordial gene (29). Four other members of this gene family are involved in the metabolism of branched-chain amino acids, lysine, and tryptophan rather than in β-oxidation (30).
The 2-enoyl-CoA moieties produced by the ACDs are hydrated to 3-hydroxyacyl-CoAs. These, in turn, undergo 2,3-dehydrogenation to 2-ketoacyl-CoAs, followed by cleavage of the thioester bond (31). This releases acetyl-CoA and completes one turn of the recursive β-oxidation cycle. The exact mechanism of these steps varies for substrates of differing chain length. The mitochondrial trifunctional protein (TFP) contains 2-enoyl-CoA hydratase, 3-hydroxyacyl-CoA dehydrogenase, and 3-ketoacyl-CoA thiolase activities for longer chain acyl-CoA substrates (32). This complex is an octamer consisting of 4α and 4β subunits. Long-chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency (LCHAD) and 3-enoyl-CoA hydratase activities reside on the α subunit, whereas 3-ketoacyl-CoA thiolase activity resides on the β subunit.
In contrast, individual proteins that catalyze these reactions for shorter chain substrates have single activities. These proteins include a short- to medium-chain 3-hydroxyacyl-CoA dehydrogenase (S/MCHAD), a shortchain enoyl-CoA hydratase (also called crotonase), and distinct medium- and short-chain 3-ketoacyl-CoA thiolases (31, 33). The substrate specificities of many of these enzymes overlap, and additional enzymes with yet different substrate optima likely exist for some steps of β-oxidation. Enzymes catalyzing several additional sets of reactions are necessary for the complete oxidation of unsaturated fatty acyl-CoA molecules including a 2,4-dienoyl-CoA reductase (34) and a Δ3-, Δ2-enoyl-CoA isomerase (26). In odd-chain (carbon number) fatty acid oxidation, the final three carbon intermediate propionyl-CoA is metabolized by propionyl-CoA carboxylase. Evidence suggests that VLCAD, ACAD9, and TFP associate with the inner mitochondrial membrane and may interact with the acylcarnitine transport and respiratory chain complexes to allow channeling of substrate directly from one enzyme to the next. Ketone bodies are produced exclusively in the liver from acetyl-CoA generated by β-oxidation (see Fig. 70.2). Hydroxymethylglutaryl (HMG)-CoA synthase forms 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) from acetoacetyl-CoA and acetyl-CoA. Acetyl-CoA and acetoacetate are then produced by cleavage of HMG-CoA by HMG-CoA lyase (19, 35). Finally, acetoacetate is reduced to D-3-hydroxybutyrate by D-3-hydroxybutyrate dehydrogenase within mitochondria (36).
Several alternative metabolic pathways become important when mitochondrial β-oxidation is impaired. Peroxisomal β-oxidation allows continued metabolism of longer-chain fats, whereas ω-oxidation in the cytosol (which proceeds from the opposite end of the fatty acid) results in the production of the characteristic dicarboxylic acids often present in these disorders. In addition, deacylation of acyl-CoA by cytosolic thioesters and conjugation of acyl-CoAs to glycine and carnitine become important mechanisms of CoA scavenging and detoxification, respectively.
PEROXISOMES
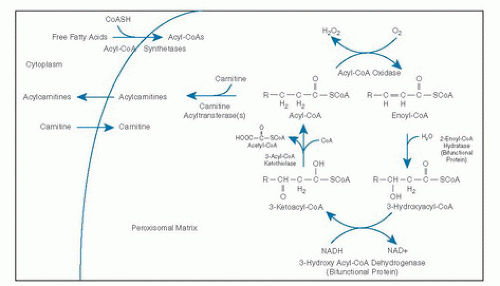
β-Oxidation in peroxisomes and mitochondria similarly involves four steps: dehydrogenation, hydration of the double bond, a second dehydrogenation, and then thiolytic cleavage. However, the β-oxidation cycle in peroxisomes differs from mitochondria in several important ways (37, 38, 39). The peroxisomal cycle results only in partial-chain shortening rather than complete oxidation of fatty acids. As a result, ATP production from peroxisomal substrates is less efficient because electrons produced by peroxisomal oxidases are donated directly to molecular oxygen for hydrogen peroxide production rather than to the respiratory chain. Carnitine plays a role in the export of chain-shortened fatty acids from the peroxisome but is not involved in fatty acid uptake.
Four peroxisomal half ATP-binding cassette (ABC) transporters have been described, including ALDP, ALDRP, PMP70, and PMP69 (40). The best characterized of these half transporters is ALDP, which is involved in the transport of acyl-CoA esters across the peroxisomal membrane. X-linked adrenoleukodystrophy (X-ALD) is caused by mutations in the ABCD1 gene coding for ALDP (40). The activation of fatty acids to fatty acyl-CoAs is accomplished by acyl-CoA synthetases in the peroxisomal membrane (41). The first step of the β-oxidation cycle in the peroxisomal matrix is oxidation by straight-chain acyl-CoA oxidase (also called palmitoyl-CoA oxidase), leading to production of an enoyl-CoA (6, 39) (Fig. 70.3). Additional oxidases can perform similar reactions using 2-methyl branched-chain acyl-CoAs and CoA intermediates of bile acids as substrates (branched-chain acyl-CoA oxidases). Because the branched-chain acyl-CoA oxidases are stereospecific, 2-methyl-CoA racemase converts (2R)-methyl fatty acids to their (2S) diastereomers for oxidation. The second and third steps of the β-oxidation cycle are carried out by a bifunctional protein complex containing enoyl-CoA hydratase and 3-hydroxyacyl-CoA dehydrogenase activities associated with the inner aspect of the peroxisomal membrane (42, 43). Peroxisomal specific 3-ketoacyl-CoA thiolases catalyze the final step of the cycle, to produce acetyl-CoA and regenerate an acyl-CoA (38, 39).
Multiple carnitine acyltransferases with different chainlength specificities catalyze the conversion of acetyl-CoA and acyl-CoAs to acetylcarnitine and acylcarnitines, thus facilitating their export from the peroxisome (41). The chain-shortening step of the synthesis of docosahexaenoic acid (DHA) (C22:6) is achieved by a single cycle of peroxisomal β-oxidation (41). Additional enzymes involved in the metabolism of unsaturated long-chain fats in peroxisomes include organelle-specific 2,4-dienoyl-CoA reductase, 3/2 enoyl-CoA isomerase, 2-enoyl-CoA hydratase, 2,5 enoyl-CoA reductase, and 3, 5/2, 4-dienoyl-CoA isomerase.
Fig. 70.3. β-Oxidation of fatty acids in the peroxisome. CoA, coenzyme A; NAD, nicotinamide adenine dinucleotide.
In addition to β-oxidation, peroxisomes are involved in several important metabolic pathways, including α-oxidation of phytanic acid and other fatty acids, ether-phospholipid biosynthesis (including plasmalogens), detoxification of glyoxylate, pipecolic acid oxidation, biosynthesis of cholesterol, and other isoprenoids and the metabolism of reactive species (6, 38).
DEFECTS OF MITOCHONDRIAL FATTY ACID METABOLISM
Defects of the Carnitine Cycle
Defects identified in this pathway include that of a specific plasma membrane carnitine transporter protein, CPT I, and CPT II, as well as the carnitine-acylcarnitine translocase (CACT). A single report of two patients with an undefined defect in fatty acid oxidation and free fatty uptake in fibroblasts has been published (44).
Deficiency of the plasma membrane carnitine transporter represents primary carnitine deficiency (23). Carnitine is freely filtered by the kidney and must be reabsorbed from the proximal tubules to preserve plasma levels. Because the transporter for carnitine is deficient in kidney as well as in muscle and liver, tissues whose carnitine content is highest, this defect results in defective renal reabsorption and reduced tissue storage of carnitine (23). This leads to a deficiency of carnitine in end organs and an impairment of long-chain fatty acid metabolism. Patients with carnitine transporter deficiency can present with severe hypoglycemia and dilated cardiomyopathy in infancy or childhood. Alternatively, they may show onset of hypertrophic cardiomyopathy, progressive muscle weakness, and muscle lipid storage with mild elevations of creatine kinase. Hypertrophic cardiomyopathy has been reported in middle-aged carriers of OCTN2 mutations. Fetal hydrops secondary to this disorder has been reported.
Multiple reports of asymptomatic, affected mothers have been identified when newborn screening of their affected or carrier children have tested positive for severely low free carnitine levels. Plasma carnitine levels are extremely low or undetectable in these children, but they rise dramatically with supplementation with pharmacologic doses of carnitine (100 to 400 mg/kg/day). Patients’ symptoms also show dramatic resolution with therapy. Outcome is likely to be good if diagnosis is promptly made and therapy is instituted. Carnitine transporter deficiency can be diagnosed by uptake studies using cultured fibroblast or direct molecular analysis of the OCTN2 gene.
Deficiency of liver CPT I has been reported. Severe disease is usual, but milder variants have been identified in geographically restricted populations. Diagnosis is based on enzymatic and mutation analysis. Severe symptoms include episodic hypoketotic hypoglycemia beginning in infancy and multiorgan system failure (45, 46, 47). Muscle and cardiac symptoms are not present. In one case, an apparently healthy girl aged 2 years and 9 months developed hepatomegaly and coma following a viral illness and died (48). Organic aciduria is not prominent in this disorder, but hyperammonemia may be present. Plasma carnitine is normal or elevated with a high free fraction. Elevated levels of creatine kinase were seen in siblings from one family. Analysis of samples from patients with CPT I deficiency has revealed normal CPT I levels in muscle but low activity in other tissues, including liver (47). Patients thus far have not responded well to therapy with carnitine, but presymptomatic treatment in subsequent affected siblings and infants identified through expanded newborn screening programs may change this observation.
Molecular analysis of patients with CPT I deficiency has identified common mutations in the CPT1A gene in the Hutterite and Canadian First Nation and Inuit populations (49, 50). Identified through newborn screening programs, affected individuals have, for the most part, been well. Patients with isolated muscle CPT I deficiency have not been reported.
Deficiency of the CACT was initially reported in newborns who had a nearly uniform poor outcome, presenting with severe hypoketotic hypoglycemia and cardiac arrhythmias or hypertrophy (51). All these infants had a grossly elevated acylcarnitine-to-free carnitine ratio, whereas dicarboxylic aciduria was reported in one. Carnitine supplementation did not appear to improve clinical symptoms. More recently, patients with a milder clinical course have been identified who responded well to modest carnitine supplementation and dietary therapy (23). Two affected siblings have been reported in which the younger sibling was prospectively treated and had not developed any sequelae 2 years later (52). These patients appear to have a higher level of residual enzyme activity than the more severely affected patients. Specific diagnosis of this disorder can be made by direct enzyme or molecular analysis.
CPT II deficiency is the most common of this group of disorders. It classically manifests in late childhood or early adulthood as episodes of recurrent exercise- or stressinduced myoglobinuria (23, 53). Episodes can be severe enough to lead to acute renal failure. Patients are typically well between episodes. They have no tendency to develop hypoglycemia. Weakness and muscle pain are reported. The characteristic diagnostic finding in these patients is a low total plasma carnitine level with increased acylcarnitine fraction and no dicarboxylic aciduria. Long-chain acylcarnitines may be elevated (23).
A more severe variant of CPT II deficiency manifesting with symptoms similar to those of CPT I deficiency has been appreciated. In these patients, the presenting symptoms were neonatal hypoglycemia, hepatomegaly, and cardiomyopathy. A severe reduction of CPT II activity was found in all tissues tested, including liver, heart, muscle, and fibroblasts, although CPT I activity was normal. Plasma carnitine levels were not increased.
Mutations in the cDNA for CPT II have been described, and expression studies of mutant CPT II alleles suggest that the level of residual function of the mutant enzyme may be responsible for determining the clinical phenotype (54). Carnitine supplementation does not benefit the severe form of CPT II deficiency (23). Familial phenotypic variation has been reported (55). A common mutation has been reported to account for half of mutant alleles in the late onset form of the disease (56). A common coding polymorphism has also been reported in the CPT II coding region that may predispose to clinical symptoms under some (unknown) circumstances. Occasional families with apparent autosomal dominant transmission of partial CPT II deficiency have been described, and at least one case appears to be related to a mutation on one CPT II allele (55, 56, 57). Why these patients exhibit symptoms is unknown, although a dominant negative effect on tetramer assembly and modifying gene effects have been postulated (58, 59).
Defects of Acyl-Coenzyme A Dehydrogenases
The first patient with VLCAD deficiency presented with ventricular fibrillation and respiratory arrest at 2 days of age and exhibited massive dicarboxylic aciduria (60). It is now clear that VLCAD deficiency can manifest with a spectrum of symptoms, including early-onset cardiac and skeletal myopathy, hypoketotic hypoglycemia, hyperammonemia, and hepatocellular failure (61). Recurrent rhabdomyolysis and myopathy beginning in adolescence have also been described (62). 3-Hydroxy-dicarboxylic acids or saturated dicarboxylic acids may be present in the urine (60, 63). Cloning of the gene for VLCAD has allowed identification of various genetic defects, but no common mutations have emerged (64). Some correlation of specific genotypes with phenotype exists, although it is imperfect. Fibroblast studies suggest that VLCAD enzyme targets different fatty acid chain lengths with different phenotypes, but this finding has not been supported in vivo (17).
One report of three cases of ACAD9 deficiency has been published (65). The first patient was a 14-year-old, previously healthy boy who died of a Reye-like episode and cerebellar stroke triggered by a mild viral illness and ingestion of aspirin. The second patient was a 10-year-old girl who first presented at age 4 months with recurrent episodes of acute liver dysfunction and hypoglycemia, with otherwise minor illnesses. The third patient was a 4.5-year-old girl who died of cardiomyopathy and whose sibling also died of cardiomyopathy at age 21 months. Mild chronic neurologic dysfunction was reported in all three patients. Defects in ACAD9 mRNA were identified in the first two patients, and all patients manifested marked defects in ACAD9 protein. Despite a significant overlap of substrate specificity, it appears that ACAD9 and VLCAD are unable to compensate for each other in patients with either deficiency.
Putative LCAD deficiency has been reported, but all the patients originally categorized as LCAD deficient subsequently were proved to have a deficiency of VLCAD instead (66). Thus, there are no known patients with bona fide LCAD deficiency.
Numerous patients with SCAD deficiency have been reported (67, 68). Clinical findings have included episodes of intermittent metabolic acidosis, neonatal hyperammonemic coma, neonatal acidosis with hyperreflexia, multicore myopathy, infantile-onset lipid storage myopathy with failure to thrive, and hypotonia. Hypoglycemia has been a rare finding in this disorder. The characteristic metabolites of ethylmalonic and methylsuccinic acids of SCAD deficiency were also detected in individuals with normal SCAD activity in fibroblasts (67). The presence of one of two relatively common variants of SCAD (625 G>A and 511 C>T) predisposes to excessive ethylmalonic acid production but it probably is not clinically important. These polymorphisms subtly affect the function of the purified proteins encoded by these variants, although both are still active (69). Few patients identified on the basis of elevated ethylmalonic acid excretion, neuromuscular symptoms, and deficient SCAD activity in fibroblasts carried two pathogenic mutations (70). The remaining patients were double heterozygous for a pathogenic mutation and the previously identified 625 G>A variation, homozygous for one of the variations, 625 G>A or 511 C>T, or double heterozygous for both.
In general, it is clear that most patients with complete SCAD deficiency identified through newborn screening have been well, whereas numerous symptoms continue to be ascribed to the deficiency in patients identified through clinical testing later in life (68, 71). The full clinical spectrum of this deficiency and the clinical relevance of common polymorphisms remain to be defined (67).
MCAD deficiency has emerged as one of the most common inborn errors of metabolism in the United States and Western Europe, and it has been extensively reviewed (17, 72, 73). The most frequent clinical presentation is one of intermittent hypoketotic hypoglycemia with onset in the second year of life (74). Mild hyperammonemia and coma may or may not be present. These findings often lead to the nonspecific diagnosis of Reye syndrome. The patient is usually well between attacks. Dicarboxylic aciduria is extensive during the attacks, but it can be undetectable by routine means when the patient is well. Similarly, microvesicular and macrovesicular hepatic steatosis, muscle weakness, and lipid excess in muscle present during the acute illness may resolve between acute episodes. Most patients who die of MCAD deficiency do so after having survived an initial episode. Thus, recurrent Reye syndrome-like episodes especially should trigger suspicion of this disorder.
Sudden death in a previously healthy child has been described in numerous cases of MCAD deficiency. This can occur as early as the first day of life, and it has been seen in a previously healthy adult who was being calorie restricted after abdominal surgery. In the appropriate age range, such deaths are often misattributed to sudden infant death syndrome (SIDS). Autopsy usually demonstrates the characteristic microvesicular and macrovesicular steatosis and should suggest the diagnosis. Analysis of the acylcarnitine and acylglycine profile from a bile specimen, as well as enzyme assay in cultured fibroblast (which may be recovered from deep tissues such as the fascia lata of the thigh up to 48 hours postmortem), may be helpful in proving it. Finally, completely asymptomatic individuals have been identified in the course of family studies of patients. The diagnosis of MCAD deficiency in asymptomatic individuals is possible by metabolite analysis of various bodily fluids (75).
Only gold members can continue reading. Log In or Register to continue