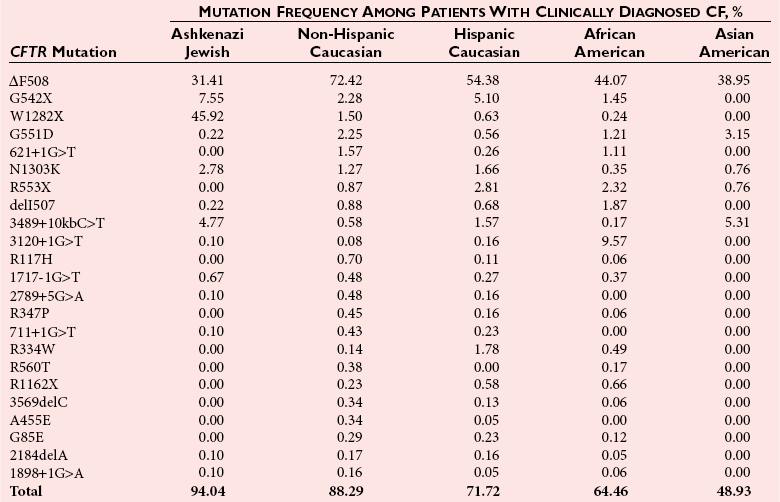
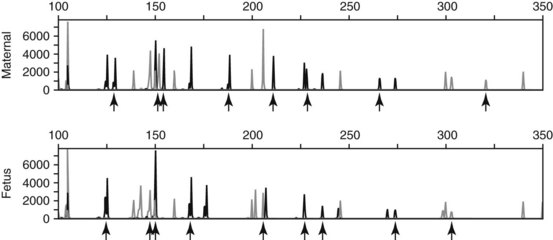
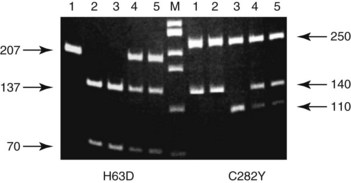
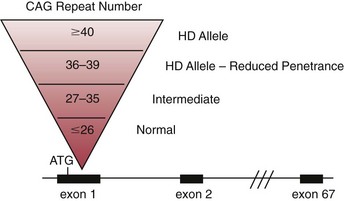
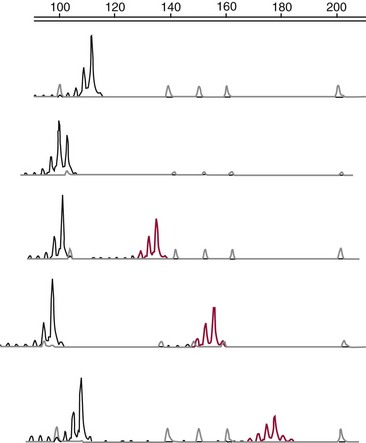
Chapter 40 Since the first description of the use of the polymerase chain reaction (PCR) for the diagnosis of sickle cell anemia in 1985, numerous applications of PCR have been described.558 The invention of PCR combined with advances in bioinformatics, the chemistry of fluorescent molecules, and the development of DNA chip and next-generation sequencing technology has revolutionized the field of human genetics. In addition, the establishment of public databases and rapid and unrestricted communication among investigators around the world via the Internet have enabled the phenomenal growth of this medical specialty. Upon completion of the Human Genome Project in 2003, the postgenomic era spawned personalized medicine, or a tailored approach to healthcare based on an individual’s genotype. The relevance of genetic variation has been explored in the areas of nutrigenomics—modulation of nutrition and nutritional requirements; vaccinomics—determination of disease susceptibility and efficacy and response to vaccines; and pharmacogenomics—evaluation of the drug response. A personalized genetic approach to medicine is most important in the area of targeted gene therapy for both inherited and acquired somatic diseases. This chapter will review deoxyribonucleic acid (DNA) testing for some of the more common inherited autosomal recessive, autosomal dominant, and X-linked genetic diseases, and will discuss testing of several mitochondrial, imprinting, and complex diseases. Because molecular testing for inherited diseases has influenced virtually every discipline of clinical medicine, the reader should refer to other chapters within this textbook for discipline-specific diseases, including inherited disorders of erythrocyte enzymes (see Chapter 23) and inborn errors of carbohydrate metabolism (see Chapter 26). In this rapidly evolving area of diagnostic testing, multiple methods can be used to achieve the same sensitivity and specificity. Similar to other areas of laboratory medicine, the method chosen by the DNA laboratory is determined by the expected volume for the test, the availability of personnel to perform the assay, and the current instrumentation within the laboratory. Cystic fibrosis (CF) (Online Mendelian Inheritance in Man [OMIM #602421]) is one of the most common autosomal recessive diseases in people of Northern European ancestry with an estimated incidence in the United States of about 1 in 2500 and a carrier frequency of about 1 in 25.5 Within other ethnic populations, the disease has an estimated incidence of 1 in 2300 Ashkenazi Jews, 1 in 8500 Hispanics, 1 in 17,000 African Americans, and 1 in 35,000 Asian Americans.5 CF is a multisystem disorder that primarily affects the pulmonary, gastrointestinal, and reproductive organs.455 However, the phenotypic expression of the disease is heterogeneous, ranging from meconium ileus and severe respiratory disease in infants to mild pulmonary symptoms and no evidence of gastrointestinal problems even in adulthood. Morbidity and mortality of the disease are most related to mucous accumulation; recurrent infection with pathogens, such as Pseudomonas aeruginosa, Burkholderia cepacia, Staphylococcus aureus, and Haemophilus influenzae; and excessive inflammation in the lung.553,559 Although it was previously considered a fatal childhood disease, the U.S. Cystic Fibrosis Foundation reported that in 2008, the median survival age among ≈23,000 patients with CF who received care through 1 of 115 care centers nationwide was 37.4 years.132 In addition, more than 45% of patients were over the age of 18.132 The increase in survival age is due to organ transplantation, improved nutrition, and new therapies.31,160,250,478,661 This success will continue with the likelihood of targeting infectious agents, mucous accumulation, and inflammation with pharmacotherapy and gene therapy to the lung.16 Further, because of the complexity and frequency of the disease, most patients receive treatment from specialized care centers.132,256 This approach provides widespread communication between healthcare providers and enables monitoring of a large population of patients with respect to treatment outcome, healthcare, and disease-specific variables. The severity and frequency of the disease led to an intensive search for the gene. The gene was mapped to 7q31 in 1985 and was subsequently cloned in 1989.365,374,536,545,633 The CF gene is large, containing 27 exons and producing a transcript of approximately 6.5 kb. It codes for the CF transmembrane conductance regulator protein (CFTR) of 1480 amino acids and a member of the ATP-binding cassette (ABC) transporter superfamily. CFTR consists of two repeated motifs, each containing six hydrophobic transmembrane domains and one hydrophilic intracellular nucleotide-binding fold connected by a highly charged regulatory domain site.536 The molecule serves as a chloride ion channel and is located within the lipid bilayer, predominantly at the apical membrane of secretory epithelial cells, where it is dependent on phosphorylation by protein kinases through activation by cyclic adenosine monophosphate (cAMP).259 In addition to epithelial chloride conductance, the molecule is involved in transport of sodium, potassium, and adenosine triphosphate (ATP) from the intracellular compartment to the extracellular surface.556,571,609 An abnormal sweat chloride concentration is considered the gold standard for the diagnosis of CF (≥60 mmol/L in childhood).223 However, some patients with CFTR mutations may have borderline (30 to 59 mmol/L) or even normal (<30 mmol/L) results.112,290 Atypical or nonclassic CF patients may have involvement of only one organ, as in congenital bilateral absence of the vas deferens (CBAVD), pancreatitis, pulmonary disease, or nasal polyps.498,685 Measuring the nasal potential difference is part of the algorithm to distinguish patients with a questionable diagnosis of CF from true CF patients.142,162,333 Thus although the diagnosis of CF can be made easily in patients with characteristic clinical features and abnormal sweat chloride concentrations, in patients with atypical presentation, mutations in the CFTR gene cannot be excluded without complete sequence analysis of the gene.434 The wide clinical diversity among patients with CF in part is the result of varying effects on the CFTR protein caused by more than 1600 mutations identified thus far in the CFTR gene.133 However, it is possible that the “nonclassic” CF phenotype is caused by factors other than mutations in the CFTR gene.255,579 Because CF is an autosomal recessive disorder, the CF patient must have two mutant CFTR alleles to develop the disease. Some mutations are “private” and unique to a family; others may be common among CF patients. Patients will be homozygous with two copies of the same mutation or will represent a compound heterozygote with one copy of one mutation and one copy of a second mutation. Predictably, the type and location of the mutation have varying effects on CFTR and affect the phenotype of the patient.6,99,455 The CFTR genotype and the clinical phenotype are most closely related for pancreatic involvement rather than for pulmonary manifestations of the disease, which appear to be more dependent on environmental factors and genetic modifiers.92,121,168,563,722 Environmental modulating factors of lung disease include second-hand smoke and varying exposure to pathogens.122,559 It is interesting to note that patients with idiopathic chronic pancreatitis have a higher incidence of CFTR mutations than the expected carrier frequency and have more severe disease.8 Pancreatitis in these patients may result from CF with a second CFTR gene mutation not detected, or could represent a complex disease resulting from a predisposing mutation in several genes.358 CFTR genotypes in infertile males with only the genital form of CF—CBAVD—usually have a mutation associated with a severe phenotype on one allele and a mutation associated with a mild phenotype on the second allele. The most frequently reported CFTR genotype in this population is the 5T polymorphism in intron 8 (corresponding to a sequence of five thymidines).112,524 The 7T and 9T alleles are more common than the 5T variant, which is observed in only ≈5% of CFTR alleles. The 5T variant affects mRNA splicing and can cause exon 9 to be deleted; without it, the chloride channel is not functional.115,144,608 An adjacent polymorphic TG dinucleotide sequence [(TG)9 to (TG)13] regulates the efficiency of mRNA splicing, with the higher number of TG repeats [(TG)13] associated with decreased efficiency of splicing.254 Thus, the 5T-(TG)12 or (TG)13 allele is more commonly associated with an abnormal phenotype than is the 5T-(TG)11 allele. The types of mutations in CFTR can illustrate the association of the genotype with the phenotype. More than half of all mutations are missense or frameshift mutations, and more than 9% occur in exon 13.133 Mutations can be divided into five classes.455 Patients with class 1 mutations have defects in protein production, and class 2 mutations are associated with defective processing of CFTR. In both cases, CFTR trafficking to the cell membrane does not occur, and both class 1 and class 2 mutations are typically associated with a severe phenotype. Class 3 and 4 mutations have CFTR expression at the cell membrane, but channel activity is reduced. Class 3 mutations can be associated with a more severe phenotype and result from defective regulation; class 4 mutations can be mild and result in diminished conduction of ion flow. Class 5 mutations are associated with abnormal splicing and thus reduced amounts of normal CFTR messenger RNA (mRNA). These mutations may be associated with a severe phenotype (621+1G>T) or a mild phenotype (2789+5G>A). The most common mutation, deltaF508, seen in about 70% of CF chromosomes in Caucasians of Northern European descent, affects processing of CFTR and prevents its trafficking to the apical membrane.111 Prevalent mutations G542X and W1282X cause premature translation termination and thus truncation of the protein.270 The frequently observed mutation G551D results in CFTR that reaches the apical membrane but improperly regulates the chloride channel.688 DNA testing for the identification of CFTR mutations is performed for a variety of reasons (Box 40-1). It is performed to confirm the diagnosis of disease in patients with equivocal sweat chloride results, or in instances when insufficient material is collected. A diagnosis of CF can be considered in the patient with only the presenting symptom of chronic pancreatitis. Alternatively, in the known CF patient, mutation analysis can be requested to help predict the prognosis because some genotype-phenotype correlations exist. At the same time, identifying the mutations segregating within a family enables preimplantation diagnosis or prenatal testing for subsequent pregnancies and carrier or diagnostic testing for other at-risk family members. Similarly, state-sponsored newborn screening programs have detected infants with CF and at the same time have identified at-risk family members and have enabled genetic testing. Most important, newborn screening allows for early diagnosis and intervention through treatment to prevent malabsorption of nutrients and to minimize lung damage from infection.578 Neonatal screening for CF is based on the immunoreactive trypsinogen (IRT) assay and is used to detect elevated levels of this pancreatic enzyme from dried blood spots. States vary in their newborn screening algorithms. In some, a positive result is followed by IRT retesting; in others, blood from the newborn is submitted for CFTR mutation analysis.497,597 A sweat chloride test is ultimately performed for confirmation of CF but may not always be effective in infants younger than 2 weeks.497 Some families referred for prenatal CF testing have no family history of CF. Rather, in these families, hyperechoic (hyperechogenic) bowel may be diagnosed in the fetus on routine ultrasonography. In one study of 209 fetuses, 7 were subsequently given the diagnosis of CF, which is about 84 times the estimated risk of CF in the general population.457 The most challenging and controversial DNA testing for CF is carrier screening for preconception and expectant couples. Carrier screening in these circumstances was recommended in October 2001 by the American College of Obstetricians and Gynecologists (ACOG) in conjunction with the American College of Medical Genetics (ACMG).14 With the heterogeneity of CFTR mutations, testing for more than 1600 mutations in this population would be a daunting task. For this reason, a core panel of 25 mutations was proposed as a minimum standard for CF carrier screening. The mutations initially proposed included those with an estimated prevalence of at least 0.1% of CF mutant alleles and a CF carrier detection rate of about 88% for non-Hispanic Caucasians.14 However, in 2004, the core panel was revised to 23 mutations with the removal of mutations 1078delT and I148T-3199del6, which are not present at a frequency representing 0.1% of all CF alleles (Table 40-1).451,607,682 The intent of the screening panel is to identify individuals at risk for classical CF, not CBAVD; thus it is recommended that 5T/7T/9T status be evaluated only in the presence of mutation R117H. In this case, the panel would allow distinction between R117H-5T and R117-7T individuals. This in turn would enable proper genetic counseling and prenatal testing options for individuals who are R117H-5T and at risk of having an offspring with CF if their reproductive partner is also a carrier of CF. Although CF is more common in the Caucasian and Ashkenzai Jewish population, the standard of care recommended by the ACOG is to make CF testing available to all preconception or expectant couples, especially because it is becoming more difficult to assign a single ethnicity to a patient to best determine her carrier risk. For CFTR mutation testing, a variety of assay platforms are commercially available and varying numbers of mutations are detected.147 Further, because the detection rate of the panel is lower in some ethnicities, laboratories serving those populations should consider supplementing this screening panel with additional mutation analysis.611 Social issues associated with population screening for this genetic disease are in part rather specific to this program (such as the enormous number of mutations in this huge gene), but many others are illustrative of issues that surround genetic testing in general. Issues surrounding this program include (1) the inability to detect CF carriers with CFTR mutations other than the 23 included in the screening panel, (2) the need for appropriate genetic counseling of patients before they consent to the analysis, (3) the need for proper understanding of the possibility of a false-negative result, (4) stigmatization of being a “carrier” of a genetic disease, (5) threats to confidentiality of results, (6) effects on health insurability, and (7) a possible increase in the number of abortions following the identification of previously unknown affected fetuses. In addition to these social and/or ethical issues, opposition to this screening panel has arisen over the set of mutations included in the panel, because those currently selected under-represent the mutations found in minorities.191,224,282,287,288 It is likely that mutations in the CFTR screening panel will continue to change as carrier screening evolves and the program is critically evaluated. It is the standard of care for all prenatal cases that DNA extracted from the fetus is tested for the presence of maternal contamination, because the presence of maternal DNA could interfere with interpretation of the results. This is best performed by using PCR amplification for highly polymorphic short tandem repeat loci coupled with capillary electrophoresis (Figure 40-1). As important as the accuracy of the laboratory results is the interpretation of these results. For carrier screening, with no family history of CF and no mutation detected, it is important for the patient to understand that a residual risk of being a carrier remains (Table 40-2).5 This is especially important for patients in ethnic groups for which the CFTR mutation detection level is reduced. Further, it is very important to know relevant family history when interpreting and reporting rest results to provide accurate genetic risk assessments regarding a residual carrier risk for a negative result, because the mutation segregating within the family could be one not detected by the test panel, and a false-negative result could occur.486 Hereditary hemochromatosis (HH) (OMIM #235200) is an autosomal recessive disorder of iron regulation that can result in excess iron deposition in otherwise healthy tissue primarily of the liver, heart, joints, pituitary gland, and pancreas. Affected individuals can absorb approximately 3 to 4 mg of iron per day compared with the normal rate of 1 to 2 mg per day, and although the average male has body iron storage of about 5 g, affected individuals can accumulate as much as 30 g. Symptoms associated with this disease occur during mid to late adulthood, but the diagnosis of HH is often delayed because early symptoms of weakness, lethargy, joint pain, and abdominal pain are nonspecific. Complications of the disease include hepatic cirrhosis, diabetes mellitus, hypopituitarism, hypogonadism, arthritis, and cardiomyopathy.615 Moreover, patients are at increased risk for hepatocellular carcinoma.366 The disease is more common in men than in women presumably because of physiologic iron loss during menstruation and pregnancy. However, other sex-related differences may be involved. The phenotypic expression of the disease appears to be dependent on both genetic and environmental factors. Although regular blood donation may be protective against HH, infection with hepatitis C, obesity-related steatosis, increased consumption of dietary iron or vitamin C (an enhancer of iron uptake), and, most notably, excessive alcohol intake can increase the likelihood of symptoms in the presence of an affected genotype.519,704 Management of the disease may include therapeutic phlebotomy, chelation therapy or erythrocytopheresis to reduce iron stores, and dietary avoidance of red meats, iron supplements, and excess vitamin C, as well as uncooked seafood (to avoid Vibrio vulnificus infection); alcohol intake should be minimal.43,80 Single-nucleotide polymorphisms (SNPs) in transforming growth factor-β1 and myeloperoxidase have been implicated as modifiers of disease progression to liver fibrosis and cirrhosis, yet the primary genetic risk factor for HFE-related disease remains the presence of a Y chromosome.704 The identification of modifier genes for this disease is an area of active interest and may involve SNPs in genes associated with iron metabolism.127 Laboratory testing for hemochromatosis most often includes determination of transferrin saturation (TS) [(serum iron/total iron binding capacity) × 100]; saturation greater than 55 to 60% is considered abnormal for men, and saturation greater than 45 to 50% is abnormal for women. Serum ferritin (SF) levels are also ordered, with serum ferritin greater than 300 µg/L abnormal for men, and values greater than 200 µg/L abnormal for women. Elevated levels of transferrin saturation and serum ferritin often lead to genetic testing. If genetic testing is positive, a liver biopsy follows to determine the amount of stainable iron and the degree of injury.332 In 1976, Simon and associates reported an association of human leukocyte antigen (HLA)-A3 and HLA-B14 antigens with idiopathic hemochromatosis, which suggested that the HH gene was located near the major histocompatibility complex (MHC) on chromosome 6p.590 Classic genetic studies confirmed linkage of the HH gene to the HLA locus, and in 1996, Feder and colleagues cloned the HH gene, HFE.196,335,389 Although HFE (6p21.3) remains the primary gene associated with abnormal iron regulation, other autosomal recessive disorders involving the genes TfR2 (transferrin receptor 2), HJV (hemojuvelin), and HAMP (hepcidin), as well as an autosomal dominant disorder involving ferroportin 1, have been reported.32 Additional genes may be associated with inherited forms of abnormal iron metabolism. The HFE gene protein, HFE, encodes a β2-microglobulin–associated protein with structural resemblance to MHC class I proteins. The HFE gene contains seven exons spanning about 10 kb, with a 2.7 kb mRNA that is highly expressed in the hepatocyte. The 40 kDa HFE protein is composed of 348 amino acids and is involved in the highly regulated hepcidin-ferroportin pathway for iron homeostasis.496 As such, when bound to transferrin receptor 1 (TfR1), HFE is silenced. However, when plasma transferrin is saturated, HFE dissociates from TfR1, making TfR1 available for binding and cellular uptake of iron. Once no longer bound to TfR1, HFE binds to TfR2 and other proteins, possibly HJV, and various bone morphogenetic proteins (BMPs) and their receptors to form an iron-sensing complex. This complex signals for the expression of hepcidin, a 25 amino acid liver hormone, using the SMAD signaling pathway. Hepcidin, now upregulated from the iron overload, binds to ferroportin, causing it to be internalized and degraded, thereby maintaining iron within the enterocytes and macrophages.496 A founder effect, suggested by linkage disequilibrium between HLA haplotypes and the HFE gene, was confirmed in two studies by the identification of homozygosity for a common mutation, G-to-A base pair substitution, in 148 of 178 (83%) and 121 of 147 (82.3%) HH patients.60,196 This mutation results in a cysteine-to-tyrosine substitution at amino acid 282 (C282Y) in HFE and disrupts disulfide bridges required for normal interaction with β2-microglobulin on the cell surface, which allows for high-affinity transferrin binding to the uncomplexed transferrin receptor.196–198 The allele frequency of this mutation is 5 to 10% in Caucasians, 1% in Hispanics, and less than 1% in African Americans.513 Although carriers of this mutation have been reported to have a twofold increased risk for acute myocardial infarction compared with noncarriers, they do not have higher transferrin saturation or ferritin than C282Y noncarriers.450,635 A second base substitution of C-to-G in exon 2 and resulting in a histidine (H)-to-aspartic acid (D) substitution at codon 63 (H63D) has been identified in a higher percentage of C282Y-negative HH patients than would be expected based on the frequency of this mutation in the population.196 This mutation is observed in 89% of HH chromosomes that do not have mutation C282Y compared with an allele frequency of 15 to 17% in Caucasian control HFE genes.196,513 HFE, with this alteration, is expressed at the cell surface, but its interaction with the transferrin receptor is altered, resulting in increased iron deposition within the cell.196,669 The frequency of mutation H63D is less in African Americans (0.026) and Hispanics (0.10).513 Mutation H63D is associated with increased risk of developing a mild form of hemochromatosis, but appears to have little effect in causing iron overload when inherited by itself (wild-type and/or H63D), which represents 2.5% of HH chromosomes, or when two copies of the mutation are inherited (H63D/H63D), which is seen in 1.4% of HH patients.419 Although many compound heterozygotes (C282Y/H63D) in the general population are asymptomatic, H63D may contribute to disease when inherited with mutation C282Y because 1 to 5% of HH patients have this genotype, although these patients display variability in liver histologic findings and iron indices.58,334,347,419,566 A third mutation in the HFE gene is associated with a mild form of hemochromatosis.30,299,461 This A-to-T mutation results in a serine-to-cysteine substitution at codon 65 (S65C) in exon 2 and is in close proximity within the gene to the previously described H63D mutation. In one study, mutation S65C was detected in 2.49% of normal controls yet was identified in 10 of 128 (7.8%) HH chromosomes that had neither C282Y nor H63D.461 Although C282Y is the primary mutation in HFE-associated HH, compound heterozygotes C282Y-H63D and C282Y-S65C have an increased risk of developing HH, thus suggesting their role in the development of HH. DNA analysis of the HFE gene is done using a variety of methods and in most laboratories includes testing only for mutations C282Y and H63D (Figure 40-2). Once HFE mutation analysis has confirmed the cause of HH in the patient, transferrin saturation (TS), serum ferritin, and DNA testing of at-risk family members can identify those who may benefit from earlier treatment and dietary restrictions.89 This is especially important because morbidity among first-degree relatives of HFE-related hemochromatosis patients is greater than in the general population.329 Because HH is a common disorder with clinical symptoms that can be prevented with easy and inexpensive early intervention, population-based DNA screening for HH may be appropriate and has been considered.93,120 Yet, although homozygosity for the HFE mutation C282Y is close to 1 in 250 in the general population, making hemochromatosis one of the most common inherited genetic disorders, significant clinical heterogeneity is observed and varies from severe disease to an asymptomatic phenotype with only abnormal iron indices. In one study of 41,000+ patients attending a health clinic in the United States, only 1% of C282Y homozygotes developed clinical hemochromatosis. However, the penetrance of C282Y homozygotes displaying iron overload–related disease in a large study of more than 31,000 people between 40 and 69 years of age was reported to be 28% in males compared with only 1% in females by the age of 65.10 Further, this study showed that patients with serum ferritin levels greater than 1000 µg/L are at increased risk for developing HFE gene-related disease compared with subjects with less than 1000 µg/L. Although clinical expression of the disease is reduced, more than 50% of homozygotes of both sexes will have elevated TS and SF levels. However, these two studies indicate wide variability of true penetrance of this disease. Thus, because of incomplete penetrance, many people with mutations will not have and will never develop iron overload.59 Inability to predict who will develop disease is the primary reason why population-based genetic screening is not done routinely in the United States. This is a topic of much debate.9 Targeted screening to adult Caucasian men of Northern European ancestry combined with TS and SF has been proposed.512 Targeted screening for family members of C282Y homozygotes has been suggested.330 In the United Kingdom, genetic testing for C282Y and H63D was determined to result in significant cost savings when incorporated into the diagnostic strategy of potentially affected patients and in testing of their offspring.128 Phenotypic measurements of transferrin saturation or serum ferritin could be more appropriate than genotype studies for population-based screening for HH, but these results can also be misleading in that iron overload can occur from a host of other conditions unrelated to hemochromatosis and mutations in HFE.699 Spinal muscular atrophy (SMA) is an autosomal recessive neurodegenerative disorder with an estimated incidence of 1 in 10,000 births and a carrier frequency of 1 in 40 to 1 in 50. The disease is characterized by progressive symmetric degeneration of spinal cord motor neurons of the anterior horn region and results in skeletal muscle weakness and atrophy.415 Wide variability in age of onset and motor function impairment has been noted in SMA, and four clinical phenotypes have been identified. Type I (OMIM #253300) is the most severe and the most common (50%); it is associated with age of onset younger than 6 months and death in early childhood. These children have profound hypotonia and no control of head movement, and they are unable to sit. Intercostal muscle weakness leads to respiratory failure and tongue fasciculation, dysphagia, and fatigue, making feeding difficult, worsening the condition, and increasing the risk of aspiration pneumonia. SMA type II (OMIM #253550) is intermediate in its severity and has an age of onset between 7 and 18 months. These children can sit and some can stand, although none can walk independently. Death typically ensues in adolescence. SMA type III (OMIM #253400) has onset of disease after 18 months and a mild phenotype with gradually progressive disease and a normal life expectancy. SMA type IV (OMIM #271150) is the mildest of all forms and is characterized by muscle weakness initially in the second or third decade of life and a normal life span. The gene for SMA, survival motor neuron I (SMN1), was mapped to 5q11.2-13.3 in 1990 and cloned in 1995.86,393,438 This gene contains 9 exons (numbered 1, 2a, 2b to 8) spanning about 28,000 bases and encodes a 1.7 kb mRNA transcript producing a 38 kDa protein composed of 294 amino acids that is widely expressed in the nucleus and cytoplasm.404 The SMN protein forms a multiprotein complex with other proteins, some of which are referred to as Gemins, and is enriched in the nucleus in size and number to form Gems.46,589 The SMN complex is essential for the biogenesis of small ribonucleoproteins into the spliceosome, a structure that is required in the nucleus for the removal of noncoding sequences during pre-mRNA splicing.505 In addition, SMN interacts with a whole host of other proteins, indicating that it plays an important role in a variety of cellular functions, including transcription and apoptosis. SMN is essential during embryogenesis in that its absence results in embryonic lethality in SMN1 knockout mice.567 It is not understood why it plays such an important role in the cell and is expressed in many tissues, yet deficiency of SMN almost exclusively affects the lower motor neurons. This would suggest that SMN may function in a role unique to motor neurons.189,717 In most cases, SMA results from homozygous deletions of SMN1. Some cases result from gene conversion of SMN1, and in less than 5% of cases, patients are compound heterozygotes with an SMN1 deletion on one allele and loss of function or point mutation on the second allele. In rare cases, a point mutation in SMN1 is present on both alleles. Reduced amounts of functional SMN in SMA patients limit the formation of the SMN complex, thereby affecting snRNP assembly and ultimately normal mRNA splicing.201,697,720 SMN1 is contained within a large inverted repeat sequence that contains the highly homologous SMN2 gene. SMN2 differs from SMN1 by only five bases; it lies in the opposite orientation and is centromeric to SMN1.415,500 It is believed that SMN2 arose by gene duplication, and that the SMN2–SMN1 configuration is unique only to primates.544 Although the five bases that differ between SMN1 and SMN2 do not affect the amino acid sequence of the protein; a C-to-T transition in exon 7 of SMN2 corresponding to codon 280 causes alternative splicing and the deletion of exon 7 in 80 to 90% of SMN2 transcripts.408 The C-to-T transition enables the binding of heterogeneous ribonucleoproteins, thereby acting as a splicing silencer element and causing the exclusion of exon 7 from the transcript.354 In addition, an SMN2-specific nucleotide in intron 7 contributes to excision of exon 7 in SMN2 transcripts.355 Thus despite the fact that most SMA patients have intact SMN2 genes, they still have disease, because most SMN2 transcripts do not contain exon 7; thus they produce negligible functional SMN protein. Without the amino acids encoded by exon 7, SMN is unstable and is unable to efficiently oligomerize to form the SMN complex.409 The SMN2 gene is in part a modifier of the severity of SMA, and its affect is based on the SMN2 gene copy number.423,622,681 Some SMN1 deletion alleles also contain an SMN2 deletion, whereas other SMN1 deletion alleles may have two or even three copies of SMN2. In patients with milder forms of the disease, three or four copies of SMN2 may be present. Increased gene copy numbers are therefore producing more SMN2 transcript, some of which will translate to functional SMN protein, thereby providing some normal SMN protein for required cellular functions. Although SMN2 copy number primarily influences SMA severity, other factors appear to be contributory. Males with identical biallelic SMN1 deletions and identical SMN2 copy numbers appear to be more severely affected than females, and the SMA phenotype is variable even within families whose members share identical genotypes.337 Phenotypic variability could be explained by recent findings of differences in DNA methylation at the SMN2 locus.278 In addition, absence of the adjacent neuronal apoptosis inhibitory protein (NAIP) gene may modify the SMA phenotype, causing more severe disease when it is also deleted.180,554,681 NAIP is deleted in 1.8% of SMA carriers, yet is homozygously deleted in 67% of SMA patients with type I disease. Treatment for SMA is mostly supportive for the management of respiratory insufficiency, nutritional deficiency, and orthopedic needs.95 Because SMN2 serves to modify the SMA phenotype, it can also provide a therapeutic target for future treatment. One approach is to reduce aberrant splicing of SMN2.367 This was successfully performed in the mouse model of SMA to increase normal SMN levels using bifunctional RNA.47,443 An alternative strategy would be to increase the expression level of SMN2 using histone deacetylation inhibitors.140 Because of the frequency and severity of the disease, therapeutic research is ongoing and many clinical trials are in progress.95 DNA testing for SMA is performed at multiple laboratories using a variety of techniques.218 Establishing diagnostic or carrier testing can be complicated by (1) the polymorphic nature of the SMN locus, with alleles containing varying copy numbers of both SMN1 and SMN2 genes; (2) the degree of homology between SMN1 and SMN2; (3) a small percentage of affected alleles with point mutations rather than deletions within SMN1; and (4) a 2% rate of de novo cases, which most frequently occur during paternal meiosis. A common diagnostic assay for SMA includes PCR amplification coupled with restriction endonuclease digestion with DraI and gel electrophoresis.646 Only amplicons derived from SMN2 will contain the restriction site; those generated from SMN1 will not. Because most SMA affected patients have SMN1 deletions involving exon 7, no bands corresponding to SMN1 are observed. Although this assay is simple and robust, it is not quantitative and cannot determine the SMN2 copy number, nor can it identify patients with point mutations within the gene. SMN2 copy number is important for prognosis and for interpretation of at-risk pregnancies. Further, this assay cannot distinguish SMA carriers from controls—a task that is important for determining recurrence risk and for distinguishing an inherited mutation from a de novo mutation. Thus in some scenarios, quantitative methods such as real-time PCR, multiplex ligation-dependent probe amplification, denaturing high-pressure liquid chromatography, fluorescent PCR coupled with capillary electrophoresis, or DNA sequencing must be used.238,312,610,674,713 Population-based carrier screening for SMA has been proposed but is not currently the standard of care.521 With prospects of therapeutic treatment for SMA and early age of onset for the disease, newborn screening for SMA has also been proposed.523 This testing would allow early identification of patients and would enable timely treatment intervention, thereby minimizing the severity of the disease. Although it is theoretically sound, widespread newborn screening for SMA is not widely available. Achondroplasia (ACH) (OMIM #100800) is the most common nonlethal form of human genetic dwarfism with a worldwide incidence of 1 in 10,000 to 1 in 30,000.308 ACH is inherited as an autosomal dominant disorder with 100% penetrance and is characterized by short-limbed dwarfism (rhizomelic form), macrocephaly, frontal and biparietal bossing, bowing of the lower extremities, and normal intelligence. The mean adult height is 131 cm for men and 124 cm for women, with a standard deviation of 5.6 and 5.9 cm, respectively. Close monitoring of patients is essential to prevent morbidity and mortality arising from complications of the disease.48 Infants with this disease can die within the first year of life from central apnea caused by compression at the craniocervical junction; two copies of the mutant allele are often lethal.241,473,502 Children undergoing surgical decompression of the craniocervical junction have decreased mortality and demonstrate improvement in neurologic function. During the first 5 years of life, affected children are at risk of death from compression of the brainstem and/or the upper cervical spinal cord. In adult patients, the leading cause of death is heart disease, and life expectancy is about 10 years less than for the general population.707 More than 90% of patients are born to parents of normal height. These patients represent sporadic cases arising as de novo mutations—a phenomenon associated with the “paternal effect” and advanced paternal age (older than 35 years).137,229 Moreover, the estimated germline frequency of the common de novo disease causing mutation may be the highest observed in human beings.692 The observed paternal effect associated with ACH has been thought to occur as the result of lifelong spermatogonial stem cell divisions, and thus an increase in production of mutant sperm as the male grows older.131 Analysis of DNA extracted from testicular biopsy samples from older subjects supports this conclusion.137 Alternatively, it has been proposed that sperm bearing ACH gene mutations may have a selective advantage in spermatogonial cells.242 With both the paternal effect and a high mutation rate, when two affected siblings born to normal-sized parents shared a 4p haplotype derived from their unaffected father, Sobetzko and coworkers suggested that two de novo, independent, sporadic events had occurred, or that paternal gonadal mosaicism was present.596 In a second reported family, recurrent affected siblings were shown to arise from paternal germline mosaicism rather than recurrent de novo mutations.469 In one unusual report, it was concluded that siblings with achondroplasia born to unaffected parents were a result of germline mosaicism from the mother.286 Although the mother had no clinical features of achondroplasia, a mutant allele was detected in the DNA extracted from her peripheral blood. In 1994, the gene for achondroplasia was mapped to the telomeric region of chromosome 4p (4p16.3) using linkage studies on multigenerational families.209,657 The fibroblast growth factor receptor 3 gene (FGFR3), mapped to this region and previously considered as a candidate gene for Huntington’s disease (HD), was evaluated as a candidate gene for achondroplasia and was reported to contain mutations in patients with achondroplasia.586,626 The FGFR3 gene consists of 17 exons and encodes an mRNA of 4000 base pairs (bp). A different set of mutations in the FGFR3 gene, causing varying effects on the function of FGFR3, have been associated with related forms of dwarfism, including thanatophoric dysplasia type I (OMIM #187601) and type II (OMIM #187601), hypochondroplasia (OMIM #146000), and a severe form of achondroplasia with developmental delay and acanthosis nigricans.274,307 The FGFR3 protein product, FGFR3, is one of four human fibroblast growth factor (FGF) receptors and contains three extracellular immunoglobulin-like domains, a single transmembrane domain, and a split intracellular tyrosine kinase domain.342 FGFR3 is a tyrosine kinase receptor that, when bound to 1 of 22 possible FGFs and heparan sulfate–bearing proteoglycans on the cell surface, induces dimerization of receptor monomers, activates tyrosine kinase activity, and promotes phosphorylation of tyrosine residues in the cytoplasmic domain, which in turn induces multiple signaling pathways, including STAT, MAPK, PLC-γ, and PI3K-AKT.308,617 Signaling through FGFR3 is complex and is not completely characterized, but FGFR3 is thought to negatively regulate chondrocyte proliferation through apoptosis and differentiation.124,145,533 Precisely which FGFs serve as ligands for FGFR3 is not known. Different proteoglycans on the cell surface, various isoforms due to alternative splicing, and different FGFs at different stages of development and at different locations of the growth plate give rise to added diversity of FGFR3 ligand binding and subsequent effects. The primary mutation in achondroplasia results in a defect in normal internalization and ubiquitination and lyosomal degradation of the mutant receptor. Thus it is retained on the cell surface and has uncontrolled and prolonged phosphorylation of FGFR3 signaling in chondrocytes.274,453 Hence, chondrocyte maturation and terminal differentiation are inhibited. As such, therapies are directed toward the increase in short stature. New therapies can be directed toward the molecular bases of disease, including (1) chemical inhibitors of FGFR3 tyrosine kinase activity, (2) antibodies that prevent binding of FGF ligands to FGFR3, and (3) targeting of regulators of various FGF-induced signaling pathways.143,263,307 In the original report identifying the FGFR3 gene as the cause of achondroplasia, 15 of 16 patients had a G-to-A transition mutation at nucleotide 1138 (c.1138G>A), and the only patient who did not have this mutation instead had a G-to-C transversion mutation at the same position (c.1138G>C).586 Both mutations result in a glycine-to-arginine substitution in the transmembrane domain of FGFR3 at codon 380. The frequency of the G-to-A transition mutation at codon 380 in achondroplasia patients is well documented and represents more than 95% of the mutation detected in this population.55,551 This base pair may be prone to mutation because a cytosine residue in a CpG dinucleotide is known to be a hot spot for transition mutations.41,64,166 If the cytosine residue is methylated (i.e., as 5-methylcytosine), it can spontaneously deaminate to thymine to introduce the change from a G : C base pair to an A : T base pair in subsequent replications of DNA. Because most FGFR3 mutations causing achondroplasia are c.1138G>A or c.1138G>C, DNA testing includes targeted mutation analysis for both mutations using a variety of techniques.218 Testing can be performed postnatally to confirm the diagnosis of achondroplasia. Prenatal DNA testing may be requested by couples in which one is affected with ACH and the risk of an affected child is 50%. Alternatively, preimplantation genetic diagnosis can be performed for ACH.12 In addition, prenatal testing can be requested by unaffected parents with a previous affected child, who may be concerned about germline mosaicism, although the risk of recurrence in these cases would be considered low. Pregnancies involving mating between two affected individuals are not uncommon. Prenatal DNA testing can be requested by these couples because they have a 25% chance of having a child homozygous for an FGFR3 gene mutation—a potentially lethal condition. This situation presents difficult choices for the couple. In one study, attitudes of affected individuals and their relatives toward termination of the pregnancy based on prenatal findings indicated that if the fetus was homozygous mutant for ACH, 40% would consider termination compared with 41% who would not; 19% were unsure of what they would do.240 In this same study, if the fetus was heterozygous for a mutation causing achondroplasia, 5% would consider termination and 86% would not; 8% were unsure what they would do. Last, if the fetus was determined to be normal, having not received a mutation from either parent to cause this disease, 90% responded that they would not consider termination of the pregnancy, but 3% would; 6% were unsure of what their decision might be. Huntington’s disease (HD) (OMIM#143100) is an autosomal dominant, late-onset neurodegenerative disorder with an incidence of about 3 to 10 per 100,000 in most populations of European origin. First described by George Huntington in 1872, the disease is progressive and is characterized by frequent involuntary, rapid movements (chorea) and dementia.320 The mean age of onset is between 35 and 44 years, but approximately 25% of patients first display symptoms after the age of 50, and about 10% of patients have juvenile HD with age of onset before 20 years.279,465 The median survival time is 15 to 20 years after onset of symptoms. In the first few years of the disease, symptoms include mood disturbances, cognitive deficits, clumsiness, and impairment of voluntary movement.279 The next stage of the disease is associated with slurred speech (dysarthria), hyperreflexia, chorea, gait abnormalities, and behavioral disturbances such as intermittent explosiveness, apathy, aggression, alcohol abuse, sexual dysfunction and deviations, and increased appetite.207 As the disease advances, bradykinesia, rigidity, dementia, dystonia, and dysphagia are present. In the late stages of HD, weight loss, sleep disturbances, and incontinence occur. In 1983, Gusella and associates reported linkage between DNA marker D4S10, on the short arm of chromosome 4, and HD, based on studies from a large kindred in Venezuela.261 Subsequently, more DNA markers were identified, and the region of the genome containing the HD gene was narrowed to 4p16.3.50,126,225,420,680 Through an international collaborative effort, and 10 years after its initial localization, the HD gene, IT15, was cloned.321 The molecular basis for HD was determined to be expansion of a glutamine-encoding CAG trinucleotide repeat; this was subsequently confirmed in a worldwide study by the identification of expanded CAG-repeat alleles in HD patients from 565 families, representing 43 national or ethnic groups.377 In this initial international study, the median CAG-repeat length was reported to be 44 in affected patients and 18 in controls. Normal CAG repeats range from 10 to 26, repeats of 27 to 35 are considered intermediate or “mutable,” repeats of 36 to 39 are associated with reduced penetrance of the disease, and repeats of 40 or greater are associated with HD (Figure 40-3). HD is one of about 20 inherited neurologic diseases associated with trinucleotide repeat instability.83,491 In HD, the number of CAG repeats is inversely correlated with age at onset of the disease. Patients with onset as early as 2 years of life have a repeat number approaching 100 or greater, and late-onset-disease patients have repeat numbers of 36 to 39.19,81,169,321,377,594 However, an autopsy confirmed that the diagnosis of HD was reported in a 65-year-old male with as few as 29 CAG repeats.364 Although the CAG-repeat number accounts for the majority of variance in age of onset for HD (≈70%), the remainder of the variance is due to modifier genes or environmental factors. Analysis of the large Venezuelean HD kindreds encompassing 18,149 individuals spanning 10 generations indicated that 40% of the remainder of the variance is due to modifier genes, and 60% to environmental factors.643 The largest genome-wide scan to identify modifying loci was performed on these Venezuelean kindreds.216 Most significant LOD scores (logarithm of the odds ratio) were identified at chromosomal regions 2p25, 2q35, 5p14, and 5q32, suggesting the physical location of candidate modifying genes. New mutations for HD (the presence of an affected individual in the absence of a family history) occur from expansion of CAG-intermediate alleles and occur almost exclusively through expansion during paternal transmission.233 Although intermediate alleles are present in about 1% of the population, it appears that flanking DNA sequences may influence the instability of these alleles by enhancing the formation of hairpin loop structures and causing replication slippage.114,234 Single sperm analysis studies have demonstrated 11% instability (9% expansions and 2.5% contractions) in CAG repeats of 30 compared with 0.6% instability (contractions only) seen in average-sized alleles of 15 to 18 repeats.392 These studies indicate that an increase in instability occurs as the repeat number increases. This concept is further supported by observations that CAG repeats of 36 showed 53% instability, and CAG repeats from 38 to 51 had instability ranging from 92 to 99%. In addition, cis-elements may play a role in CAG-repeat instability in IT15.678 Warby and colleagues identified a common haplotype (A) of 22 tagged single-nucleotide polymorphisms (tSNPs) that was more common on HD (95%) and intermediate (83%) alleles in patients of European origin than on normal (53%) alleles from the same population. Onset of symptoms occurs at progressively younger ages in successive generations of affected families, a pattern called anticipation. Anticipation is explained by meiotic expansion of the unstable CAG repeat during transmission by the affected parent, resulting in an even higher CAG-repeat number in the offspring and an earlier age of onset. In addition, although 69% of affected father-child pairs show expansion, only 32% of affected mother-child pairs demonstrate expansion. Further, <2% of maternal expansions result in a change of >5 repeats, whereas up to 21% of paternal transmissions increase by >7 repeats.376 The affected parent in most cases of juvenile-onset HD is the father. However, the largest reported CAG-repeat number of ≈130 occurred via maternal expansion of 70 CAG repeats.467 An increase in the CAG-repeat number is also associated with more rapid progression of disease and greater neuropathologic severity in the striatum.213,508 However, it is interesting to note that homozygotes with two expanded CAG-repeat alleles do not have more severe disease than heterozygotes.377,464,690 The HD gene protein, huntingtin (htt), consists of 3144 amino acids with a molecular mass of ≈349 kDa, is ubiquitously expressed in all tissue, and predominantly resides in the cytoplasm with lesser amounts in the nucleus.148,262,302,627,655 With universal distribution, it is not known how and/or why the primary phenotype is neuronal loss. In neurons, htt is associated with synaptic vesicles and microtubules and is abundant in dendrites and nerve terminals. Huntingtin interacts with multiple proteins, indicating its function in intracellular trafficking and cytoskeletal organization, as well as in endocytosis, apoptosis, and transcription regulation.* Mutant HD alleles are effectively transcribed and translated, but as a result of the increase in glutamine residues, the protein is misfolded.564 Thus, abnormal folding may result in aberrant protein-protein interaction of mutant htt with any of its protein partners and could contribute to the pathogenesis of HD. In addition, neuronal mitochondrial dysfunction may play a role in the disease process.85,675 Further, aggregates of fragmented mutant htt form neuronal nuclear inclusion bodies (IBs), which may be toxic to the cells.156 Recent data, however, suggest that IBs may result from a normal cellular process to handle abnormal and toxic proteins, and that their formation is facilitated by proteasomal chaperones.448,550 HD remains an incurable disease; however, various therapeutic strategies have been proposed and/or attempted in animal models. Such strategies include allele-specific silencing of mutant IT15 mRNA; small molecules targeted toward mutant htt; and cell transplantation therapy.360,620,719 DNA testing for HD is performed with the use of PCR so that the exact CAG-repeat number can be determined.321 Soon after the initial report, it was discovered that the accuracy of determining the CAG-repeat number by PCR was compromised by inclusion of a polymorphic CGG repeat immediately downstream from the CAG repeat. Because the CGG repeat was contained within the amplified sequence, the length of the amplicon could be altered and overestimated.20,555 Upon this discovery, a new primer pair was identified that flanked the CAG repeat, yet excluded the problematic polymorphic CGG repeat and provided accurate assessment of the CAG-repeat number.679 However, in contrast to earlier years, when PCR involved the use of 32P-labeled primers for this and similar assays, currently the most common method involves the use of PCR with fluorescently labeled primers and capillary electrophoresis664 (Figure 40-4). Guidelines for diagnostic testing by clinical laboratories with reference to standardization and interpretation have been developed.518 Besides the technical and interpretive difficulties associated with HD testing, many ethical issues exist as well, primarily as they relate to presymptomatic testing (Box 40-2). The first policy statement on ethical issues related to genetic testing for HD was adopted in 1989 at a joint meeting with representatives from the International Huntington Association and the World Federation of Neurology.325 At that time, the gene had not yet been cloned, and predictive testing was performed using linkage studies; the at-risk patient was quoted the likelihood of inheriting the mutant allele. These tests were less than perfect and provided, at best, results in only 60 to 75% of families. Moreover, the possibility of recombination allowed erroneous predictions to occur.280,463 In other families, living affected members were not available, or markers were not informative. Once the gene had been cloned and direct mutation analysis was possible, risk assessments were reversed in a small percentage of patients.11 Box 40-2 Ethical Issues Associated With Presymptomatic DNA Testing for HD Patients must be 18 years of age. The decision to proceed with testing must be voluntary and informed. Genetic counseling regarding benefits/pitfalls of testing is required. A support partner is needed for the patient for counseling and the testing process. Diagnosis of Huntington’s disease (HD) in the family should be confirmed by DNA testing before presymptomatic testing. Psychiatric assessment of patient is necessary before testing. Follow-up genetic counseling is recommended after delivery of results. Discrimination by insurance carriers or employers may occur following completion of testing. Prenatal testing of fetus is controversial; preimplantation diagnosis is available.
Inherited Diseases
Diseases with Mendelian Inheritance
Cystic Fibrosis
Hereditary Hemochromatosis
Spinal Muscular Atrophy
Autosomal Dominant Diseases
Achondroplasia
Huntington’s Disease
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree