Introduction
The inflammatory rheumatic diseases form a group of disorders that are highly variable in their phenotypic expression. However, they have in common the presence of localized and/or systemic inflammation, which results in characteristic connective tissue and internal organ damage. Among these diseases, the specific clinical and pathologic features of each disorder likely reflect the initiating and propagating stimuli that determine the specific tissues targeted, and the inflammatory effector mechanisms that predominate.
Although the spectrum of inflammatory rheumatic diseases is broad, some general principles provide a framework within which to discuss the pathophysiology of all. One of the most useful constructs is a kinetic one, which focuses on disease initiation, propagation and flares. It is useful for discussion of both acute and chronic diseases. Understanding the stimuli and mechanisms responsible for each of these phases among the different rheumatic diseases permits a deeper insight into these fascinating and complex syndromes.
Overview of Inflammatory Rheumatic Diseases
The initiating force of acute diseases (eg, gout, immune complex vasculitis) is often exogenous and clearly recognizable (eg, crystal deposition, new medication, systemic bacterial or viral infection). The disease is self-limited due to the success of the inflammatory response in removing the offending initiating stimulus (eg, crystals in gout; bacterial antigen or drug in immune complex vasculitis; Figure 24–1). Despite resolution of the acute episode, flares may occur upon reexposure to the initiating stimulus.
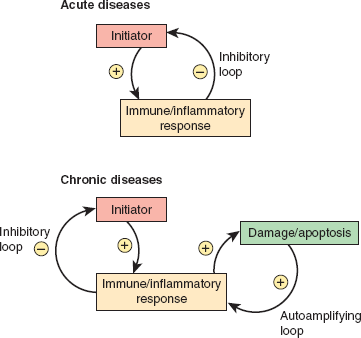
The initiating force in chronic diseases (eg, systemic lupus erythematosus [SLE], rheumatoid arthritis) is often remote and no longer recognizable once the unique disease phenotype becomes fully established and the diagnosis clear. Propagation of the disease typically occurs as a result of an autoimmune response, inducing a self-amplifying cycle of damage. Conditions leading to the initiation of chronic autoimmune diseases occur rarely, but once a disease is established, flares are frequent. This circumstance probably reflects the abundant capacity of the immune system to “remember” previously encountered antigens and to respond to them with greater vigor when encountered again, even at lower concentrations (Figure 24–1).
Different tissues are affected in various diseases (eg, specific synovial joints in gout and rheumatoid arthritis; skin, joints, kidney, serosal surfaces, nervous system, and blood cell lines in SLE).
Pathogenesis of Inflammation
The nature of tissue damage and joint injury is determined in part by the inflammatory and immune effector functions that predominate. In addition, the pathologic features of the chronic inflammatory disorders reflect the combination of inflammatory damage and the consequences of healing.
Recruitment and activation of specific subsets of inflammatory and immune cells are essential determinants of the pathologic features. In this regard, the role of activation of regional blood vessel endothelium by proinflammatory cytokines (eg, tumor necrosis factor [TNF], interleukin [IL]-1) must be emphasized. Several cytokines induce the expression on endothelial cells of ligands for the adhesion-promoting receptors of inflammatory cells (integrins and selectins) and allow neutrophils and monocytes to adhere to the vessel wall in the inflamed area and migrate into the underlying tissues.
Distinct classes of immune effector function are activated depending on the pattern of cytokines that predominate during initiation of the inflammatory response. For example, some cytokines (eg, IL-12) produced by infected monocyte-macrophages skew the lymphocyte response toward TH1 cells (which generate the TH1 cytokines IL-2, interferon-γ, and TNF) that are associated with activation of macrophage killing functions and protection against invading intracellular pathogens. In contrast, the presence of IL-4 during the initial response induces the differentiation of TH2 lymphocytes, which generate TH2 cytokines (eg, IL-4, IL-5, IL-6, and IL-10). These cytokines have their predominant function in the activation of B cells and antibody generation. A new subset of helper T cells that develop in the presence of the cytokines TGF-γ and IL-6 has recently been described. These cells are termed TH17 cells because of their characteristic secretion of IL-17. They appear to be critically involved in granulocyte recruitment, protection against certain types of bacteria, and generation of chronic inflammation and autoimmunity.
Although significant overlap exists, specific pathologic features tend to accompany the different cytokine patterns (eg, granulomas for TH1 versus immune complex disease for TH2). In addition, significant recent data point to an important role for type I interferons in inducing novel pathways of monocyte differentiation in patients with SLE that enhance responses to self-antigens.
The classical complement pathway is activated when antibody binds to its specific antigen. Activation of the complement cascade induces inflammatory cell recruitment and activation (with all the consequences mentioned later) as well as other features of the acute inflammatory response (eg, increased capillary permeability).
Although myelomonocytic cells (neutrophils and macrophages) have numerous effector pathways that function to rid the host of foreign invaders, some of these effector mechanisms can damage healthy tissue if released in large amounts. These include free radical species generated during the respiratory burst as well as a variety of secretory products contained in the granules of these inflammatory cells. Important granule contents include a variety of proteases such as cathepsins, elastase, and collagenase. These products are liberated into the extracellular medium in the inflammatory locus, where they accumulate and may have damaging effects on normal connective tissue. In addition, numerous proinflammatory mediators released in this environment (including TNF, IL-1, IL-6, prostaglandins, and leukotrienes) attract further inflammatory cells to the area and amplify the potential to generate tissue damage if the inflammatory response is not adequately quenched.
Numerous studies have emphasized the roles of the complement pathway and immunoglobulin Fc gamma receptors (FcγR) in the activation of myelomonocytic cell effector function that result in tissue damage. For example, Fc receptors play a critical role in generating the pathologic picture characteristic of immune complex–mediated diseases (see below). Clinical conditions in which this situation might arise include drug reactions, serum sickness, and infections (infective endocarditis, streptococcal skin and pharyngeal infections, and others). Autoimmune diseases are characteristically antigen driven, but in this case, the humoral response is directed against self-antigens (eg, nucleosomes [composed of DNA and histones] in SLE). Under conditions leading to the liberation of significant amounts of self-antigen from host tissue (cell damage or death), immune complex formation, Fc receptor binding, and complement activation may result.
The consequences of immune complex formation and deposition are similar whether caused by foreign antigens or self-antigens. Notably, immune complex–mediated renal disease and vasculitis that occur in several murine models of SLE are completely absent in the FcγR knockout mouse.
Certain T lymphocytes (ie, CD8+ T cells) are capable of killing target cells. When target cell destruction exceeds the capacity for renewal, impaired tissue function can result. As with other lymphocyte functions, this effector function is activated only on ligation of the T-cell receptor by a specific peptide (bound within the cleft of a major histocompatibility complex [MHC] molecule). On recognition of antigen on the surface of a target cell, cytotoxic T lymphocytes induce the death of those cells, using several distinct mechanisms. One prominent mechanism involves the Fas-Fas-ligand (FasL) pathway, whereby FasL present on activated lymphocytes binds to the Fas receptor on target cells and activates target cell apoptosis. The second prominent mechanism involves the release of cytotoxic T-lymphocyte secretory granules. These granules contain at least two distinct classes of proteins. One, called perforin, allows water, salt, and proteins (including the second class of granule protein, the granzymes) to enter the target cell cytoplasm through mechanisms that still remain unclear. The granzymes, a family comprising several proteases, target a number of critical cellular substrates and activate the process of apoptosis (programmed cell death) within the target cell.
The destruction of antibody-coated target cells by natural killer cells is called antibody-dependent cellular cytotoxicity (ADCC) and occurs when the Fc receptor of a natural killer (NK) cell binds to the Fc portion of the surface-bound antibody. The cytotoxic mechanism involves the release of cytoplasmic granules containing perforin and granzymes into the cytoplasm of the antibody-coated cell (similar to cytotoxic T-lymphocyte–mediated killing, described previously).
This mechanism has been implicated in autoantibody-mediated syndromes, in which the autoantigen resides at the cell surface or relocates to this site after an insult. An example of this is the photosensitive skin disease that occurs in patients with SLE who possess the Ro autoantibody. On exposure to ultraviolet light, the Ro antigen is released from keratinocytes and binds to their surface. Circulating Ro antibodies bind the antigen at this site, with induction of FcR-mediated effector pathways.
In response to inflammatory mediators (including cytokines) and T cells, cells in tissues ordinarily unrelated to the immune response can alter their form and function to support (and in some cases drive) a chronic inflammatory response. This mechanism has been recently described in myositis (see below), where the inflammatory and autoimmune response is focused to areas of ongoing damage and regeneration.
Checkpoint
1. What is the hallmark of the rheumatic diseases?
2. What three kinetic features account for the specific clinical and pathologic characteristics of the different rheumatic diseases?
3. What six inflammatory effector mechanisms account for the inflammation seen in the rheumatic diseases? Give an example of a disease that illustrates each principle.
Pathophysiology of Selected Rheumatic Diseases
Gout is the classic example of crystal-induced inflammation of synovial joints. It is a common condition, presenting in approximately 4% of the adult American population, and it is approximately 3 times more common in men than women. Deposition of monosodium urate crystals in the joint space leads to episodes of severe acute joint pain and swelling (particularly in the great toe, midfoot, ankle, and knee). These episodes tend to resolve completely and spontaneously within a week even in the absence of therapy. If not properly treated, however, this acute, self-limited form of the disease can evolve over many years into a chronic, destructive pattern resulting in more frequent and sustained periods of pain and resultant joint deformity. Accumulations of urate crystals elsewhere in the body can lead to subcutaneous deposits called tophi.
The critical initiating factor in gout is the precipitation of monosodium urate crystals in synovial joints. This occurs when body fluids become supersaturated with uric acid (generally at serum levels >7 mg/dL). Indeed, the degree of hyperuricemia correlates well with the development of gout, with annual incidence rates of about 5% for serum uric acid levels more than 9 mg/dL. Increased levels of serum uric acid result either from underexcretion (90% of patients) or overproduction (10%) of uric acid. A decreased glomerular filtration rate is the most frequent cause of decreased excretion of uric acid and may be due to numerous causes (see Chapter 16), but, regardless of etiology, impaired renal function is clearly related to the occurrence of gout. Diuretic administration is also a frequent cause of decreased excretion of uric acid. Overproduction defects can result from primary defects in the purine salvage pathway (eg, hypoxanthine phosphoribosyl transferase deficiency), leading to an increase in de novo purine synthesis and high flux through the purine breakdown pathway. Diseases causing increased cell turnover (eg, myeloproliferative disorders, psoriasis) and DNA degradation (eg, tumor lysis syndrome) are secondary causes of hyperuricemia.
Although the concentration of monosodium urate in joint fluid slowly equilibrates with that in the serum, formation of crystals is markedly influenced by physical factors such as temperature and blood flow. The propensity for gout to involve distal joints (eg, great toes and ankles), which are cooler than other body parts, probably reflects the presence of local physical conditions at these sites remote from the body core that favor crystal formation.
Monosodium urate crystals are not biologically inert. Their highly negatively charged surfaces function as efficient initiators of the acute inflammatory response. The crystals are potent activators of the classic complement pathway, generating complement cleavage products (eg, C3a, C5a) that are strong chemoattractants for neutrophil influx (Figure 24–2). The crystals also activate the kinin system and in that way induce local vasodilation, pain, and swelling. Phagocytosis of crystals by synovial macrophages activates the inflammasome (a complex of proteins that sense certain intracellular stressors and activate IL-1 maturation) and stimulates the release of proinflammatory cytokines (eg, IL-1, TNF, IL-8, PGE2). These products increase adhesion molecule expression on local vessel endothelium to facilitate neutrophil adhesion and migration and are also potent chemoattractants for neutrophils. Neutrophils also amplify their own recruitment by releasing leukotriene LTB4 upon phagocytosis of urate crystals (Figure 24–2).
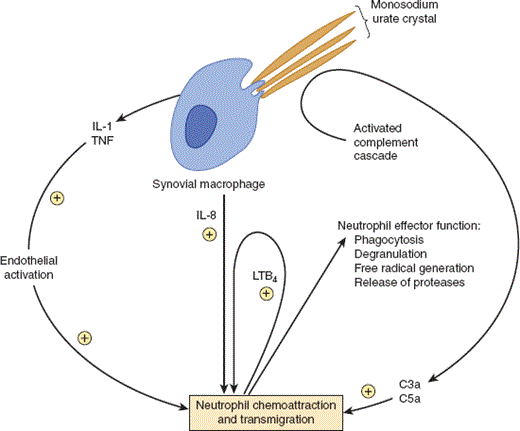
The intense inflammatory response in gout typically resolves spontaneously and completely over the course of several days, even without therapy. This down-modulation of the inflammatory response is a typical feature of acute inflammation, whereby the inflammatory response itself successfully removes the proinflammatory stimulus (Table 24–1). Numerous mechanisms appear to be responsible: (1) efficient phagocytosis of crystals, preventing activation of newly recruited inflammatory cells; (2) increased heat and fluid influx, altering local physical and chemical conditions to favor crystal solubilization; (3) coating of crystals with serum proteins, rendering the surface of the crystals less inflammatory; (4) secretion of a variety of anti-inflammatory cytokines (eg, TGF-β) by activated joint macrophages; and (5) phagocytosis of previously activated apoptotic neutrophils by macrophages in the joint, altering the balance of cytokines secreted by these macrophages in such a way that secretion of proinflammatory cytokines is inhibited while anti-inflammatory cytokine secretion is enhanced.
|
Thus, gout represents an excellent example of an acute inflammatory response initiated by a proinflammatory force. The response is acute, highly focused, and self-limited rather than self-sustaining and associated with little tissue destruction in the acute phase. Flares of disease represent recurrence of crystals in a proinflammatory form in the joints. Myelomonocytic cells and humoral factors (eg, cytokines and the complement and kinin cascades) are critical mediators of the acute syndrome.
Podagra—severe inflammatory arthritis at the first metatarsophalangeal joint—is the most frequent manifestation of gout. Patients typically describe waking in the middle of the night with dramatic pain, redness, swelling, and warmth of the area. Flares of gout typically produce one of the most intense forms of inflammatory arthritis. The toes and, to a lesser extent, the midfoot, ankles and knees are the most common sites for gout flares. Gout attacks frequently occur in circumstances that increase serum uric acid levels, such as metabolic stressors leading to increased DNA or adenosine triphosphate (ATP) turnover (eg, sepsis or surgery) or dehydration. Agents that reduce prostaglandin synthesis (eg, nonsteroidal anti-inflammatory drugs), reduce neutrophil migration into the joints (eg, colchicine), or decrease the activation of myelomonocytic cells (eg, corticosteroids) reduce the duration of a gouty flare.
Gouty arthritis can be diagnosed by examination of synovial fluid from an actively inflamed joint under a polarizing microscope. Monosodium urate crystals can be seen as negatively birefringent needle-like structures that extend across the diameter of and are engulfed by polymorphonuclear neutrophils.
Firm, irregular subcutaneous deposits of monosodium urate crystals may occur in patients with chronic gout and are referred to as tophi. Tophi most often form along tendinous tissues on the extensor surfaces of joints and tendons as well as on the outer helix of the ear. Such tophi may extrude chalky material, containing urate crystals, onto the skin surface that can be viewed for diagnostic purposes under polarized microscopy.
In some patients, the total body burden of uric acid increases greatly over years; deposits of monosodium urate crystals occur at multiple joint sites. This may result in a persistent but more indolent inflammatory arthritis associated with remodeling of the thin synovial membrane into a thickened inflammatory tissue. Destructive and irreversible joint deformities resulting from bone and cartilage erosions often develop in this circumstance. Renal tubular injury and nephrolithiasis can also develop under these conditions.
Therapy for acute gouty arthritis consists of agents that decrease inflammatory cell recruitment and activation to the involved joints. In contrast, prevention or prophylaxis of recurrent attacks of gouty arthritis requires chronic therapy to decrease serum uric acid levels into the normal range, where dissolution of crystals is favored. Several agents are available that can accomplish this purpose. These include uricosuric agents (eg, probenecid), which enhance excretion of uric acid into the urine, allopurinol and febuxostat, which impede uric acid synthesis by inhibition of xanthine oxidase (a critical enzyme in the uric acid synthetic pathway), and pegloticase, which converts uric acid to allantoin, an inactive and soluble metabolite that is readily excreted by the kidneys. Conceptually, the xanthine oxidase inhibitors, allopurinol and febuxostat, are most appropriate for the treatment of uric acid overproduction (10% of patients), the uricosuric agent, probenecid, for the treatment of uric acid underexcretion (90% of patients), and pegloticase for the rare cases of refractory gout. However, agents that decrease uric acid production can be used for therapy of hyperuricemia irrespective of cause and are often more convenient in terms of dosage regimens.
Checkpoint
4. What physical factors other than uric acid concentration influence crystal formation in gout?
5. What are some proinflammatory products released by synovial macrophages upon phagocytosis of urate crystals?
6. Suggest five reasons why the intense acute inflammatory response in gout typically resolves spontaneously over the course of several days even in the absence of therapy.
7. What are three metabolic conditions that can precipitate a gout flare?
8. Name three chronic sequelae of recurrent gout flares.
Immune complex vasculitis is an acute inflammatory disease of small blood vessels that occurs in the setting of ongoing antigen load and an established humoral (antibody) immune response. Tissues affected include the skin (leukocytoclastic vasculitic rash), joints (inflammatory arthritis of small and medium-sized synovial joints), and kidney (immune complex–mediated glomerulonephritis).
Antigens are frequently derived from exogenous sources, including infections (eg, streptococcal skin infections, hepatitis B virus) and numerous drugs (especially antibiotics). An intense inflammatory response to such antigens accounts for one of the names (“hypersensitivity vasculitis”) given to this disorder. Release of endogenous antigens in the setting of an autoimmune response (eg, SLE; see later discussion) may similarly initiate the vasculitic process.
Any antigen that elicits a humoral immune response may give rise to circulating immune complexes if the antigen remains present in abundant quantities once antibody is generated. Immune complexes are efficiently cleared in most circumstances by the reticuloendothelial system and are rarely pathogenic. Their pathogenic potential is realized when circulating immune complexes are deposited in the subendothelium, where they set in motion the complement cascade and activate myelomonocytic cells. The propensity for immune complexes to deposit is a function of the relative amounts of antigen and antibody and of the intrinsic features of the complex (ie, composition, size, and solubility). The solubility of immune complexes is not a fixed property, because it is profoundly influenced by the relative concentrations of antigen and antibody, which generally change as an immune response evolves. For physicochemical reasons, soluble immune complexes formed at slight antigen excess are not effectively cleared by the reticuloendothelial system and are of a size that allows them to gain access to and be deposited at subendothelial and extravascular sites (Figure 24–3). When antibody is present in excess, immune complexes are rapidly cleared by the reticuloendothelial system and deposition does not occur.
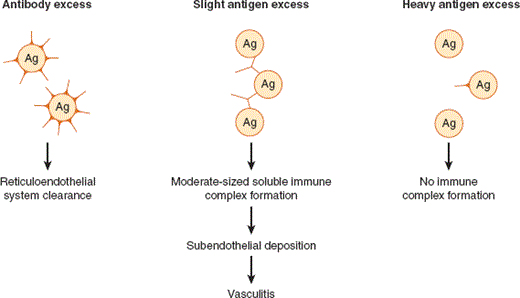
Thus, if foreign antigens (eg, drugs or infectious organisms) induce an antibody response in the setting of slight antigen excess, significant numbers of immune complexes of the appropriate size are formed and they may then be deposited in small vessels in various target organs (in skin, joints, kidney, blood vessel walls) where they activate several effector pathways (eg, FcR receptor, classic complement cascade) and where they may lead to the characteristic skin rashes (eg, palpable purpura), arthritis, and glomerulonephritis, which are the hallmarks of small-vessel vasculitis. As the immune response progresses and titers of specific antibody rise, or as the offending agent is removed, complexes are more effectively cleared, leading to resolution of the vasculitis.
A classic example of the altered pathogenicity of immune complexes at various antigen-antibody ratios is serum sickness. (Penicillin-induced hypersensitivity vasculitis represents a similar example.) When serum products from animals (eg, horses) are injected into humans for a therapeutic purpose (eg, as once was used for passive immunization against snake venom), the foreign serum proteins stimulate an immune response, with antibodies first appearing approximately 1 week after injection. Soon thereafter, immune complexes appear, followed by the development of fever, arthritis, rash, and glomerulonephritis, consistent with deposition of soluble immune complexes and myelomonocytic cell activation at multiple tissue sites. As the antibody titers rise, immune complexes are no longer formed at great antigen excess but approach the zone of equivalence and then the zone of antibody excess. The latter complexes are effectively cleared and thus lose their pathogenicity as the immune response evolves. Provided that antigen administration is not sustained, the inflammatory disease will resolve spontaneously as those immune complexes that were deposited early (during the soluble phase) are cleared. Such significant clinical effects of immune complexes usually occur only when the initial antigen load is great (eg, a large bacterial load or drug administration).
Affected tissues are all highly enriched in small blood vessels, which are the target of injury in this syndrome.
A common clinical presentation of immune complex–induced vasculitis in the skin is palpable purpura, which appears as red or violaceous papules. Cutaneous immune complex vasculitis seldom causes severe pain or tissue breakdown and only rarely leads to long-term injury (see Chapter 8).
The most common pattern of joint involvement with immune complex disease is that of a severe, rapid-onset and self-limited symmetric polyarthritis. As the immune complexes undergo phagocytosis and are cleared, the immune response remits unless further waves of immune complexes are deposited.
Glomeruli are beds of small blood vessels in the kidney where immune complexes are likely to be deposited. Acute immune complex glomerulonephritis causes proteinuria, hematuria, and formation of red blood cell casts, due to disruption of the glomerular basement membrane caused by subendothelial complex deposition. In cases of extensive immune complex–mediated injury, immune complex vasculitis can cause oliguria and acute kidney injury.
The most effective treatment for immune complex vasculitis is elimination of the inciting antigen (eg, by discontinuation of an offending drug). Medications that reduce the degree of activation of myelomonocytic cells (eg, corticosteroids) are also helpful.
Contrast between Immune Complex Vasculitis, Granulomatosis with Polyangiitis [Formerly Wegener Granulomatosis], & Polyarteritis Nodosa
The vasculitides are a diverse group of inflammatory syndromes characterized by inflammatory destruction of blood vessels. However, not all forms of vasculitis are caused by immune complex deposition. This fact is highlighted by the current classification system for the systemic vasculitides, which segregates diseases on the basis of the size of the blood vessel involved (Table 24–2), by the presence of circulating autoantibodies, and by the histologic presence or absence of immune complexes.
| Vessel Size | Examples | Epidemiology and Demographics |
|---|---|---|
| Small vessel | Immune complex mediated; Henoch-Schönlein purpura | Common, evanescent. Predominantly in children, relatively common compared with other autoimmune conditions |
| Medium vessel | Polyarteritis nodosa | Rare; ~5 cases per million |
| Large vessel | Giant cell arteritis | Only in patients older than 50 years; ~100 cases per million |
It is useful to contrast the clinical and pathophysiologic features of immune complex vasculitis (see prior discussion) with those of the “pauci-immune” vasculitic processes, which include granulomatosis with polyangiitis [GPA] and polyarteritis nodosa. The clinical hallmarks of GPA include granulomatous inflammation of the upper airway (eg, sinusitis) and lower airway (eg, trachea, lungs), as well as a necrotizing vasculitis involving the kidneys and many other organs. Although immune complex deposition is not a prominent feature in the pathophysiology of GPA, a specific group of antibodies highly specific to this disease may play an important propagating role. These “ANCA” antibodies [antineutrophil cytoplasmic antibodies], directed against components situated within neutrophil cytoplasmic granules, may bind to and activate neutrophils at the interface of the plasma and vessel wall and cause them to degranulate and damage vascular walls at these sites.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


