
Figure 7-1. Leukocyte recruitment. 1. Circulating leukocytes express integrins in a low-affinity conformation. 2. Exposure to activated endothelium leads to rolling, which is mediated by L-selectin and P-selectin on the neutrophil and E-selectin on endothelium. 3. Leukocyte exposure to cytokines released by macrophages phagocytosing pathogens induces a high-affinity integrin conformation integrins. Tight leukocyte–endothelial adhesion involves integrin engagement with counterligand expressed on the endothelium. 4. Subsequent exposure to chemokines leads to diapedesis, which is further mediated by the family of β1 and β2 integrins. (Adapted from Abbas AK, Lichtman AH. Cellular and Molecular Immunology. 5th ed. Philadelphia, PA: Saunders; 2003.)
Once tightly adhered, neutrophils must diapedese between endothelial cells and across the basement membrane to arrive at the focus of inflammation. Platelet/endothelial cell adhesion molecule 1 (PECAM-1) and integrin-associated protein are integral to transmigration (Fig. 7-1).13,14,16,17 PECAM-1 is concentrated along the intercellular junctions of endothelial cells, and both leukocyte and endothelial PECAM-1 appear to be essential for neutrophil and monocyte diapedesis. Other candidate receptors include the β1 integrins, or very late antigens (VLA), which possess affinity for many constituents of the extracellular matrix, including laminin, fibronectin, and collagens, the β3 family of integrins including the glycoprotein (gp) IIβIIIα and the vitronectin receptor.4 Further “directions” for migration to the focus of inflammation are delivered by the concentration gradients of chemotactic factors, including complement C5a, IL-8, LTB4, and the bacterial product N-formylmethionyl-leucyl-phenylalanine (fMLP).18–20
The clinical significance of even minor derangements in any aspect of this process is evident in the disease leukocyte adhesion deficiency, characterized by complete absence of CD18, and therefore all β2 integrins. Patients usually succumb to recurrent skin and mucosal infections within the initial 10 years of life.4
Phagocytosis
Microbial elimination commences upon first encounter with a foreign pathogen. It is facilitated by opsonization, a process in which microbes are coated by immune globulins and/or complement, which subsequently bind to their respective cell surface receptors, FcγRs and Mac-1.21–23 Neutrophils constitutively express low-affinity immune globulin receptors FcγRII and FcγRIII and can be induced to express high-affinity FcγRI by incubation with IFNγ or cross-linking β2 integrins.5,24 Complement-dependent phagocytosis is mediated by interactions between the leukocyte Mac-1 receptor and the complement opsonin iC3b.
Once engaged, FcγRs are phosphorylated on tyrosine residues within an immunoreceptor tyrosine activation motif (ITAM) by the Src family kinases.24 These phosphorylated sites serve as docking regions for a variety of proteins, in particular Syk. The importance of Syk is underscored by the observation that mice deficient in Syk are incapable of ingesting IgG-opsonized particles. A series of enzymes are subsequently activated including phosphoinositol 3-kinase, phospholipase C (PLC), and protein kinase C. Ultimately, the actin cytoskeleton undergoes rearrangement and the local plasmalemma is remodeled in the formation and sealing of the phagosome.4,24
This immature phagosome undergoes a series of maturation steps, whereby it acquires the machinery necessary for the killing and disposal of internalized microorganisms. Alterations in cytosolic calcium concentration induce the fusion of secretory vesicles and granules containing the microbicidal armamentarium with the immature phagosome.24 Proteins effected by calcium concentration and that may govern phagosomal maturation include synaptotagmins, actin, calmodulin, and the Src family of kinases.24 The soluble N-ethylmaleimide-sensitive-fusion-protein attachment protein receptor (SNARE) proteins are thought to assist in fusion by engaging cognate receptors on the target membrane and approximating the two membranes. Antibodies to the SNARE 5, syntaxin 6, and SNAP-23, inhibited exocytosis of azurophilic and specific granules, respectively.24

Figure 7-2. Integrin Signaling. Integrins comprise a large family of cell surface receptors that are composed of 2 subunits, a and b, and are activated by dimerization. The cytoplasmic tails are devoid of enzymatic activity, and hence, signal transduction is effected by adapter proteins that connect the receptor to the cytoskeleton, cytoplasmic kinases, and transmembrane growth factor receptors. As integrins bind the extracellular matrix they become clustered and associated with the cytoskeletal proteins talin, paxilin, and vinculin and signaling complexes. Actin stress fibers form, which increase integrin clustering. Ultimately, focal adhesion kinase (FAK) is recruited via interactions with talin and paxillin or with the b integrin subunit. Subsequent autophosphorlyation on tyrosine 397 provides a binding site for the Src homology 2 (SH2) domain of Src. The Src kinase phosphorylates a number of focal adhesion components including paxillin and tensin and pl30CAS, a docking protein that recruits Crk, which can subsequently activate proximal elements in the JNK cascade of the MAPK family. FAK may also be phosphorylated by Src on tyrosine 925, creating a binding site for the complex of the adapter Grb2 and Ras guanosine 5′-triphosphate exchange factor mSOS. These interactions also lead to activation of MAPK cascades, and ultimately the induction of a variety of genes. (Adapted from Giancotti F & Ruoslahti E. Integrin Signaling. Science 1999:1028-1032)
Neutrophil Granules and Secretory Vesicles
Neutrophils exhibit an oxygen-dependent “respiratory burst” pathway that generates toxic oxygen derivatives and an oxygen-independent pathway that utilizes toxic proteinases.25 These two microbicidal arms are compartmentalized into four distinct granules or vesicles (Table 7-2).26 They are mobilized in a hierarchical fashion in response to gradual elevations in the intracellular calcium level, which parallels the current needs of the cell.27 They also contain adhesion molecules and important inflammatory mediators that help further orchestrate the involvement of other cells.
Primary, or azurophil (affinity for the dye azure A), granules target the destruction of phagocytosed organisms (Table 7-2). The myeloperoxidase (MPO) within these granules generates hypochlorous acid (HOCl) from products generated by nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and also imparts the characteristic greenish color of pus.9,27 Other major constituents include (1) α-defensins, cytotoxic proteins that scaffold into transmembrane pores within the microbial cell wall and perturb the maintenance of vital transmembrane gradients, and (2) bactericidal/permeability-increasing (BPI) protein, which binds to gram-negative organisms and induces rearrangement of membrane lipids and inhibits growth.27–30 Elastase cleaves constituents of the extracellular matrix, including proteoglycans, collagen (types I, III, IV), and fibronectin and, of course, elastin.31 Azurocidin is chemotactic for monocytes and stimulates LPS-induced release of IL-6 and TNFα from monocytes.4,27
Table 7-2 Neutrophil Granules and Secretory Vesicles

Specific granules are rich in antimicrobial substances that are released extracellularly (Table 7-2).26 Lactoferrin, by sequestering iron, retards bacterial growth and can bind bacterial cell membranes and induce irreversible membrane damage and lysis.27 Phospholipase A2 (PLA2) participates in the degradation of bacterial membrane phospholipids. Lysozyme, present in all granules, is a cationic antimicrobial peptide that cleaves peptidoglycan polymers of bacterial cell walls. Gelatinase and collagenase are other extracellular matrix degrading enzymes.27 These granules also possess receptors for a variety of extracellular matrix proteins and cell surface ligands (i.e., β2 integrin Mac-1) that mediates firm adhesion to the endothelium. In addition to the mechanical function of cellular anchorage, engagement of these receptors with their respective counterligands induces phenotypic alterations such as degranulation and enhanced ROS production.4,27
Gelatinase granules contain matrix metalloproteases, zymogens that upon proteolytic activation degrade the interstitial matrix including collagens, fibronectin, proteoglycans, and laminin; this may facilitate neutrophil extravasation and migration (Table 7-2). They too are a source of cell surface adhesion molecules.4,27,30
Secretory vesicles contain many of the cell surface adhesion molecules essential for leukocyte recruitment. Their membranes are dense with the β2 integrins LFA and Mac-1, the complement receptor CR1, the LPS receptor CD14, and the FcγRIII. Through fusion with the plasmalemma, the cell surface is enriched with these receptors, which facilitates firm neutrophil-endothelial engagement and the capacity to respond to a variety of stimuli.4,27 Not surprisingly, because of the essential nature of leukocyte recruitment, secretory vesicles possess the lowest threshold for release and thus released earliest, followed by gelatinase, specific, and azurophil granules (Table 7-2).27
Table 7-3 Major ROS and Their Metabolism

Oxidative Burst and Oxidant Metabolites
The generation of toxic oxygen metabolites and ROS is the cardinal characteristic of the neutrophil, though is also a defensive quality of the monocyte/macrophage. Their essential antimicrobial properties are equally destructive to host tissues and implicated in the pathophysiology of many inflammatory disorders (Table 7-3). Thus, a delicate balance must be maintained between ensuring elimination of the offending pathogen and minimizing host damage.
A free radical is any species possessing one or more unpaired electrons. They are categorized under the broader term ROS, which encompasses all molecules capable of radical formation. Despite a very brief existence (10−11 to 10−6 seconds) extensive damage may occur through the induction of free radical chain reactions. Species underlying both physiologic and pathophysiologic inflammation include the following: the superoxide anion (O2•−), the hydroxyl radical (•OH), hydrogen peroxide (H2O2), the singlet oxygen, and the reactive nitrogen intermediates nitric oxide (NO) and peroxynitrite (ONOO−) (Table 7-3). Counterintuitive to their role in cell destruction, recent evidence also supports a role of ROS in intracellular signal transduction.32 Thus, in addition to their antimicrobial properties, ROS can modulate the immune response by activating inflammatory cells and inducing proinflammatory cytokine secretion.33,34
2 Implicit with the capacity for pathogen elimination is the potential for destruction of host tissues. Hence, numerous regulatory mechanisms provide temporal and spatial control of the ROS production. The NADPH oxidase complex itself exists in a disassembled state, and only upon cell activation and the need for ROS production are the subunits approximated and enzymatic function enabled.35 In addition, high plasma and tissue concentrations of proteinase inhibitors provide continuous surveillance and systemic control. However, such regulation is incomplete as evidenced by diseases such as rheumatoid arthritis, chronic obstructive pulmonary disease, and autoimmune vasculitis, which are the consequence of damage due to neutrophil-derived products.35
NADPH oxidase is a heteromeric complex composed of six subunits: flavocytochrome b558, the electron transporting apparatus comprises gp91(phox) and p22(phox); the cytosolic complex p40(phox), p47(phox) and p67(phox); and the oxidase factor rac-2 (Fig. 7-3).35 Microorganisms or high concentrations of chemoattractants bind to cell surface receptors and initiate oxidase activation, heralded by phosphorylation of p47(phox). Cytochrome b558 (gp91 and p22), which exists within the plasmalemma and the membranes of specific granules and secretory vesicles, is recruited by phagocytosis and granule fusion. Phosphorylation of p47(phox) induces a conformation change that enables its incorporation within the membrane, wherein it facilitates the translocation of rac2 and p67(phox), and stabilizes the association of this cytosolic complex with cytochrome b558, thereby rendering the complex functional.35
At the redox center of the oxidase an electron is transferred from NADPH to oxygen, thereby generating superoxide.36
O2 + e–=O–2
Other mechanisms of superoxide production include uncoupling of xanthine dehydrogenase system, uncoupling of mitochondrial and endoplasmic reticulum (ER) electron transport chains, and nonenzymatic reactions such as autoxidation of hemoglobin.37,38
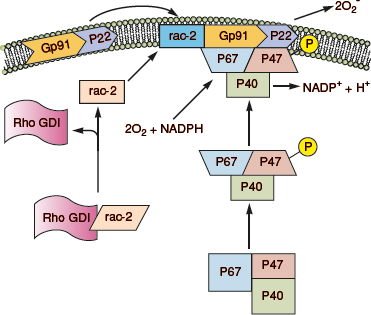
Figure 7-3. NADPH oxidase assembly. In the resting neutrophil, the cytochrome subunits gp91 and p22 are tightly bound in the membrane. P47(phox), p67(phox) and rac-s complex are in the cytosol. On activation, GDI releases rac-2, and p47(phox) becomes phosphorylated. This causes translocation of rac-2, p47(phox), and p67(phox) to the membrane and complex formation with the cytochrome components, thereby completing the assembly of the active oxidase. (Redrawn from Burg ND, Pillinger MH. The neutrophil: function and regulation in innate and humoral immunity. Clin Immunol 2001;99(1):7–17.)
Superoxide is relatively weak and of low bactericidal potency. However, its membrane permeability and role as a reactant in reactions yielding highly toxic products confers upon it a high potential for cellular and tissue damage.36
Superoxide can spontaneously or enzymatically (superoxide dismutase) dismutate into hydrogen peroxide.35,36

It can also be converted to the more potent hydroxyl radical through the metal-catalyzed Haber–Weiss reaction.35,36

or through the Fenton equation35,36

Under physiologic conditions, lactoferrin found in neutrophil specific granules provides the iron catalyst for the Haber–Weiss reaction. In the Fenton reaction, superoxide or other biologic reducing agents such as lactate or ascorbate donate electrons to generate the ferrous ions required to react with hydrogen peroxide to produce the hydroxyl radical.35,36,39
The hydroxyl anion is highly reactive and induces DNA strand breaks and base hydroxylations leading to adenosine triphosphate (ATP) depletion and gene mutations. It can attack lipid side chains of membrane phospholipids to form hydrogen peroxide and lipid hydroperoxides in a process called lipid peroxidation. These products can disrupt membrane function, serve as substrates for the production of cytotoxic aldehydes, or uncouple calcium-ATPase and increase cytosolic calcium concentration. Recent data also support a mechanism by which oxidation of critical sulfhydryl residues on the ryanodine receptors induces an “open” configuration and a leak of intracellular ER calcium into the cytosol.40 This elevation of cytosolic calcium activates calcium-dependent proteases and phospholipases that propagate cellular damage.39
Superoxide can react with nitric oxide to produce peroxynitrite (ONOO–) and hydroxyl radical.36,41

The hemoprotein MPO yields the potently bactericidal HOCl from the reactants chloride and H2O2. HOCl oxidizes amino acids, nucleotides, and hemoproteins, can activate neutrophil collagenases and permit unabated elastase injury by inhibiting α1-antitrypsin, and contribute to hydroxyl radical and singlet oxygen production.33,39,42 Though short-lived, subsequent reactions with secondary amines generate secondary chloramines, which are equally toxic but much more stable. These metabolites can oxidize similar cellular components. They can combine with halide anions to generate toxic free halides or with taurine chloramines that induces membrane attack complex (MAC) complement formation.33,39,42
In light of the pivotal role of MPO during inflammation and pathogen elimination, it is surprising that MPO deficiency is common and relatively benign. Though MPO-deficient neutrophils show early depressed bacterial killing, bactericidal function normalizes within 60 minutes.43 It is hypothesized that though bacterial killing is impaired, post-phagocytosis-oxidase–dependent neutrophil apoptosis is normal, resulting in appropriate regulation of the inflammatory response.43
Singlet oxygen, a highly reactive and extremely short-lived species, is formed by an input of energy to O2 that reverses the spin direction of one of the outermost unpaired electrons away from a parallel spin. It is produced during reactions of the MPO–H2O2–halide system and is a potential product of superoxide dismutation and the Haber–Weiss reaction. It is highly electrophilic, reacting with compounds containing electron-rich double bonds and may react with membrane lipids to produce peroxides.39
The destructive potential of these ROS for both host and pathogen necessitates a mechanism of continuous tight spatial and temporal regulation. The oxidase complex itself exists spatially disaggregated; only upon cellular activation are its constituents assembled and enzymatic function restored (Fig. 7-3).35,43 As elegant is the mechanism by which to control ROS production, so too are the measures employed to eliminate these products when no longer needed. Superoxide, the proximal reactant necessary for many of the ROS generating reactions, is removed by both spontaneous and enzymatic (superoxide dismutase) dismutation to H2O2 (Fig. 7-4). H2O2 is subsequently reduced to oxygen and water by catalase.36,39 In the extracellular environment, this function is performed by GSH peroxidase, a selenium-dependent enzyme that reduces H2O while oxidizing reduced GSH to its oxidized form. The utilization of N-acetylcysteine, a reducing agent that restores GSH, reduces hepatocellular injury in an animal model of warm liver ischemia/reperfusion.32 It has also been shown to reduce contrast-induced nephropathy in patients undergoing imaging procedures requiring the use of iodinated contrast.44 Mechanisms for preventing hydroxyl radical-induced tissue damage include the binding of transition metal ions by albumin, ceruloplasmin, haptoglobin, lactoferrin, and transferrin.33,36,39 Taurine is a scavenger for HOCl. Other antioxidants that may assist in controlling the reaction include vitamins E (tocopherol) and C. Vitamin C has many antioxidant properties, including the ability to regenerate α-tocopherol. It can prevent activation of neutrophil-derived collagenase and is a powerful scavenger of HOCl, superoxide, singlet oxygen, and hydroxyl radicals. Carotenoids have long double bonds to attract and sequester free radicals. Uric acid is a powerful scavenger of water-soluble radicals such as HOCl and singlet oxygen. It can also bind copper and iron ions to suppress hydroxyl radical formation. Stress proteins or heat-shock proteins (HSPs) are induced by oxygen radicals and ischemia and may play a role in defense. Furthermore, heme oxygenase-1 (HO-1) catalyzes the cleavage of heme to biliverdin, which is subsequently converted to bilirubin, an efficient free radical scavenger.33,36,39
All of the aforementioned participants of the NADPH oxidase are vital for health, as evidenced by those who suffer from chronic granulomatous disease (CGD).35,43 These patients have deficient superoxide production and experience ineffective inflammatory reactions to infection. They commonly suffer from repeated bacterial infections (pneumonia, cutaneous abscesses and hepatic and perihepatic abscess, and osteomyelitis) by organisms that are catalase positive (Staphylococcus aureus). Organisms that produce large amounts of peroxide are less of a threat as the neutrophils can utilize bacterial peroxide to produce toxic metabolites. The use of prophylactic antibiotics and IFNγ has reduced the frequency of serious infections in this patient population.35,43

Figure 7-4. Scavengers of ROS. (Redrawn from Klebanoff SJ. In: Gallin JI, Snyderman R, eds. Inflammation: Basic Principles and Clinical Correlates. 3rd ed. Philadelphia, PA: Lippincott Williams & Wilkins; 1999:723.)
Neutrophil Extracellular Traps and NETosis
The arsenal of antimicrobial products and mediators possessed by neutrophils to fulfill the tasks of killing and eliminating pathogens is vast and impressive. However, another distinct antimicrobial “weapon” was recently described, in which neutrophils extrude a lattice of chromatin and histones to entrap and then kill invading pathogens. The characteristic feature is the presence of neutrophil nuclear DNA in the extracellular space. These structures were entitled neutrophil extracellular traps (NETs), and the process of NET formation called NETosis. Attached to this DNA backbone is an array of microbicidal agents that are typically compartmentalized inside the azurophilic, specific and gelatinase granules: neutrophil elastase (NE), cathepsin G, and MPO. Distinct from apoptosis, the process does lead to cell death, though clearly contributes to pathogen control. It is conserved in various vertebrates, insects, and even plants, and several microorganisms exhibit mechanisms of resistance to NETs, both of which may highlight the importance of this process. Recent data suggest that excessive NETosis or ineffective NET clearance may underly diseases or syndrome characterized by excessive inflammation, including autoimmunity.45–47
Pathogens (i.e., bacteria, fungi, viruses, and protozoa) and their subcellular components, such as LPS, M1 protein, and fMLP induce NET formation. Similarly, sterile insults, inflammatory stimuli (e.g., PAF, IL-8, TNFα, NO), and chemical compounds (PMA, ionomycin) all can stimulate NETosis. The process begins with engagement of the aforementioned stimuli with TLRs, Fc receptors, cytokine, and complement receptors located on the neutrophil cell surface. Subsequent calcium release stimulates PKC activity and assembly of functional NADPH oxidase, leading to ROS and NO production. ROS is/are requisite for NET formation, as antioxidant scavengers (e.g., N-acetylcysteine) attenuate the process. Interestingly, individuals with CGD, a disease characterized by insufficient ROS production, inefficiently produce NETs. The chromatin undergoes decondensation followed by mixing of nuclear and cytoplasmic components with granulate antimicrobial contents. Plasma membrane rupture ensues with the release of chromatin decorated with granular proteins into the extracellular space.45–47
NETs are able to trap almost all pathogens, including those too large to be phagocytosed. Indeed, it is proposed that trapping by NETs is one of the most important functions that limits microbial spread. The antimicrobial functions of NETs are mediated by the microbicidal compounds accompanying the chromatin: NE, MPO, histones, cathepsin G, proteinase 3, lactoferrin, calprotectin, and antimicrobial peptides such as defensive or the cationic LL37. However, the histones H1, H2A, H2B, H3 and H4 themselves also possess powerful antimicrobial effects.
As fundamental as NETosis is for microbial defense, excessive formation or ineffective clearance of NETs is proposed to lead to several pathologic conditions. During acute lung injury a high concentration of stimulating factors promotes NETosis and the release of NE, LL-37, and ROS, which may exacerbate injury. In cystic fibrosis, the high concentrations of DNA due to NETosis are thought to underlie the high mucus viscosity.45–47 Several autoimmune diseases are also characterized by perturbed NETosis, and more than 70% of NET components are potent autoantigens. Small vessel vasculitis is characterized by antineutrophil cytoplasmic antibodies reactive against PR3 and MPO. Externalization of IL-17 in the extracellular traps of neutrophils and mast cells is now thought to be central to the pathogenesis of psoriasis. In systemic lupus erythematosus (SLE), an imbalance between NET formation and clearance may underlie the systemic tissue damage that occurs. In fact, decreased NET degradation correlates with renal disease. NETs may also activate complement, thereby amplifying the disease. Furthermore, the neutrophils from patients with various autoimmune diseases appear more prone to NETose.45–47
NETs may also serve as a link between inflammation and thrombosis. They can provide a stimulus and scaffold for thrombus formation by promoting platelet and RBC adhesion and by concentrating effector proteins and coagulation factors. Activated endothelium produces compounds that, upon contact with neutrophils, stimulate NETosis, which in turn, promotes endothelial damage.45–47
Regulation of Neutrophil Activity
In addition to the previously described mechanisms for controlling the inflammatory response of neutrophils, there is substantial evidence supporting the role of apoptosis in halting and resolving the neutrophil-derived inflammation (Box A).48 Within 90 minutes of phagocytosis, over 250 genes are induced, of which more than 30 encode proteins integral to at least three distinct apoptotic pathways.48 These observations suggest that the mechanism inducing apoptosis is initiated quite proximal in the inflammatory cascade, in fact, just subsequent to phagocytosis. The timely execution of a controlled cell death program in human PMNs is essential for preventing damage to healthy tissues and for the resolution of infection. Furthermore, evidence suggests that the phenotype of other immune cells, including monocytes and macrophages, is altered after encountering and phagocytosing apoptotic neutrophils. Thus, apoptotic neutrophil particles may function to downregulate the inflammatory function of neighboring cells. In some circumstances of infection or trauma, neutrophil apoptosis may be delayed, which might contribute to prolonged or a failure to resolve inflammation.
The inflammatory capacity of neutrophils is also transcriptionally regulated. Genes encoding proinflammatory mediators or signal transduction molecules such as receptors for IL-8, IL-10, IL-13 are downregulated early after activation and decrease rapidly after the initiation of apoptosis.48 In addition, regulating oxidative stress and ROS achieve high priority as the genes involved in glutathione and thioredoxin metabolism and heme catabolism are upregulated as is the production of reduced glutathione.48 Hence, activation-induced apoptosis in neutrophils stimulates self-directed regulation, an event that likely facilitates removal of neutrophils by macrophages. As aforementioned, removal of apoptotic neutrophils by activated macrophages also appears to serve a role in modifying their function and halting the inflammatory response.
Mononuclear Phagocytes
Monocytes circulate for about 1 to 2 days, whereafter, they constitutively hone to a particular tissue (i.e., lung, peritoneum) to differentiate into macrophages possessing a phenotype specific to the resident tissue (dendritic cells [DCs] of the skin, kupffer cells of the liver) (Table 7-1).4,49,50 Resident macrophages are typically found at interfaces with blood (liver and spleen) and with lymph, where they can readily detect, ingest, and destroy invading organisms.39 Mononuclear cells function as antigen-presenting cells (APC) in T cell–mediated adaptive immune responses, presenting antigen in the appropriate context to effector T cells. They provide service integral to both innate and adaptive immune responses. Evidence also supports their role in providing an “alarm” both locally and systemically through the release of intracellular proteins (i.e., high-mobility group box 1 [HMGB1]) expressing DAMP that can function as a danger signal (see below).
Recruitment
Monocytes are recruited and emigrate to foci of inflammation utilizing similar mechanisms of adhesion and diapedesis as described for neutrophils (Fig. 7-1). PAF, C5a, the CC chemokines, regulated on activation, normal T cell–expressed and secreted (RANTES), MIP-1α, and chemokines of the membrane cofactor protein (MCP) family are potent monocyte-macrophage chemotaxins.51,52 The selectin family of adhesion receptors mediates the initial tethering of monocytes to endothelial cells.18 Firm adhesion to the endothelium involves the interactions of β1 and β2 integrins on monocytes with the endothelial adhesion molecules ICAM-1 and VCAM-1.18
Phagocytosis
Phagocytosis involves the IgG receptor (FcγR) and the receptor for the complement factor C3b. Terminal sugar patterns on microbial surfaces also allow recognition by macrophages for nonspecific phagocytosis through the mannose lectin pathway.22 However, phagosomal maturation differs from that which occurs in the neutrophil. Monocytes and macrophages have an endocytic pathway targeting the phagosome to a lysosome.24 After endocytosis of a receptor–ligand complex, the contents of a vesicle are targeted to an early endosome; the ligand and receptor dissociate, and the receptor is then recycled to the cell surface.24 This early endosome undergoes a series of maturation steps in which it is acidified (pH 5.5 to 6.0). This acidification is requisite for optimal protease and hydrolase activity involved in pathogen killing. It may also be integral for phagosome maturation as titrating the acidity inhibits phagosome–lysosome fusion.24 Ultimately, the endosome fuses with a lysosome, which is characterized by its extreme acidity (pH <5.0) and elevated concentration of proteases.24 Lysosomes are the terminal destination of phagocytosed material targeted for degradation.
After fusion, MPO released into phagosomes can react with hydrogen and halides to yield toxic hypohalous acids, superoxide anion, hydrogen peroxide, and hydroxyl radical (Table 7-3). Macrophages may also use peroxidase generated by adjacent neutrophils, eosinophils, and monocytes and acquired through endocytosis to generate these ROS. In addition to supporting the inflammatory response, macrophages also play an important immunoregulatory role in inflammation by scavenging apoptotic neutrophils at sites of inflammation.39
Activation
IFNγ derived primarily from T cells is the primary activator of macrophages.4,50,53 Optimal macrophage activation requires both interferon IFNγ and a sensitizing agent, both of which can be provided by activated T lymphocytes. CD40 ligand on T cells can bind CD40 on macrophages to sensitize the cell. Alternatively, membrane-associated TNFα or lymphotoxin from lymphocytes can activate macrophage TNFα synthesis and thereby sensitize the macrophage to IFNγ.39 IL-10 promotes monocyte maturation and macrophage differentiation.54 Other activators include GM-CSF, TNFα, IL-1, and LPS.
Activated monocytes and macrophages can produce approximately 100 different products, including GM-CSF, M-CSF, G-CSF, IL-1, TNFα, G-CSF, and NO.39,55 Mononuclear phagocytes are important sources of chemoattractants such as IL-8, PAF, and leukotriene B4 (LTB4) that recruit neutrophils and other leukocytes. Their release of HMGB1 and other DAMP molecules serves as an endogenous “danger” signals to other immune cells in the local environment. These bind to various PRR to alter cell function. However, systemic release of HMGB1 may also be causally related to mortality in such inflammatory states as sepsis and trauma.56,57 The respiratory burst and subsequent production of toxic ROS mirrors that of neutrophils.
Antigen Presentation
T cells recognize only those antigens associated with surface major histocompatibility complex (MHC) molecules. MHC class I molecules are expressed on all nucleated cells, whereas MHC class II molecules are restricted to APC. After phagocytosing pathogen, mononuclear cells process and display antigen to T cells, and in doing so, initiate the development of the adaptive response (i.e., antibody formation). There is evidence that the HSP receptor CD91 may also participate in this process (see below). This processed antigen is presented on the APC cell surface in the context of MHC molecules that are specifically recognized by T-cell receptors (TCRs) and essential for T-cell activation. CD4+ T cells, or helper T cells (TH), recognize antigen coexpressed with MCH class II molecules and induce B-cell differentiation into either memory or antigen-specific antibody-producing plasma cells. These TH cells can also induce macrophage production of NO, ROS, and other inflammatory mediators. CD8+ cytotoxic T lymphocytes (CTL) recognize antigen in the context of MHC class I molecules and induce target cell lysis; they destroy host cells infected with intracellular pathogens or cells of malignant potential.58 Activated mononuclear phagocytes release IL-12, a potent stimulus for TH cells and the production of inflammatory cytokines, and elaborate IL-15, the function of which mirrors that of IL-2.59,60
The three professional APC are DCs, macrophages, and B cells. DCs are a specialized APC, which process and present antigen to naïve T cells. Monocytes stimulated with GM-CSF, IL-4, or IL-13 differentiate toward DC. Maturation of the DC requires TNFα or LPS stimulation.39 Epidermal Langerhans cells, after encountering antigen, migrate through the lymphoid organs and differentiate into mature DC. DC cells are particularly effective at presenting viral antigen. They present antigen in the context of both MHC I and MHC II, and thereby induce both a TH1 and a TH2 response, respectively. DC can also present antigens derived from apoptotic cells in the context of MHC I.61–63
Macrophages present antigenic peptides from ingested pathogens that persist in the phagosomes. These peptides, usually of bacterial origin, are expressed in conjunction with MHC II molecules. B cells, by contrast, bind specific soluble molecules (insect toxins, venom, and allergens) via immunoglobulin. This is endocytosed, processed, and presented on surface MHC II.39,63–65
Lymphocytes
B, T, and NK cells comprise this lineage of inflammatory cells (Table 7-1). B and T cells are central to the adaptive immune response, whereas NK cells lack antigen specificity and primarily function during innate immune response. NK cells are the first line of defense against many viral infections. The loss of surface expression of MHC class I molecules as occurs with virus-infected cells serves as a target for NK cells.39,66 Alternatively, NK cells bind cell-bound antibody and participate in antibody-dependent cell cytotoxicity. Cells targeted by either mechanism are induced to undergo cell death.
B Lymphocytes
B cells, though of bone marrow origin, attain full maturation within extramedullary sites such as lymph nodes, the spleen, and the mucosal lymph nodules of tonsils and Peyer patches. With activation, B cells differentiate into antibody-producing plasma cells, which through the elaboration of antibody, aid the neutralization of viruses and bacterial toxins, and facilitate opsonization for phagocytosis and complement activation.4 Activation requires antigen binding to cell surface receptors and stimulation by TH cell–derived cytokines; they do not need the assistance of APCs. Polyclonal B-cell activation can occur in a T-cell–independent mechanism if the antigen has a large repeating polymeric sequence.39,67
T Lymphocytes
Development of T lymphocytes begins within the marrow and is completed in the thymus. The final population profile is determined by apoptotic processes of both positive and negative selection.68 Any protein or antigen of host origin is presented by APCs, and thymocytes reactive to these self-proteins are deleted.69 Alternatively, expansion of cell lines recognizing foreign, or rather nonself antigen, occurs through positive selection. IL-7 provides the stimulation for proliferation and differentiation of developing T cells. Ultimately, two lines of mature cells, CD4+ and CD8+, will develop.69
Lymphocytes, the smallest of the leukocytes, constitute approximately 20% of circulating leukocytes.4 Most circulating lymphocytes are T cells, and 60% of those are CD4+, a marker of a TH phenotype. The other 40% are CD8+, called cytotoxic T cells, TC. The normal ratio of CD4+/CD8+ is 2:1.4 Lymphocytes continuously recirculate through lymph nodes, spleen, lymphatics, lymph nodules, and blood, providing continuous surveillance. Encounter with a particular antigen initiates activation toward an effector T cell. Activation of a T cell requires bind to its specific antigen plus a costimulatory signal provided by the interaction between costimulatory molecules on the APC and their cognate receptors on the T cell.39
Naïve T cells circulate continuously between blood and lymphoid organs, making contact with many APCs and the epitopes of the antigens they express.70 Initially, lymphocytes enter the cortical region of lymph nodes by migrating across the high endothelial venules, a process mediated by the selectin family of receptors. L-selectin, which is found constitutively on all lymphocytes, binds sialyl Lewis carbohydrate on the endothelium.39 For example, L-selectin on lymphocytes binds GlyCAM-1 on the high endothelial venules in lymph nodes. In mucosal tissues, L-selectin and endothelial MAdCAM-1 guide emigration. Migration across the endothelium requires integrins, in particular LFA-1 and its interaction with ICAM-1 and ICAM-2 on endothelial cells.71 Most lymphocytes are carried back to the blood by the efferent lymphatics. If a T lymphocyte recognizes its specific antigen on the surface of an APC, it remains for several days, then returns to the blood as an armed effector T cell.39
Adhesion molecules mediate many of the transient interactions between T cells and APCs that are required for the T cell to sample each antigen it encounters. Lymphocyte LFA-1 can bind the APC in a loose, reversible fashion by any of the ICAM molecules on APCs. If a match between T cell and antigen is found, conformational changes in LFA-1 greatly increase its affinity for ICAM-1 and ICAM-2 to stabilize the interaction. The T cell can then proliferate and differentiate into an effector cell. Effector T cells lose surface expression of L-selectin and no longer circulate through lymphoid tissue. Instead they express VLA-4, an integrin, which binds vascular endothelium at sites of infection, and the cell is retained at the focus. Effector T cells have increased LFA-1 and CD2 adhesion molecule expression that facilitate tight binding to target cell.72,73
Antigen binding in the appropriate context provides the signal for clonal expansion and differentiation of T cells into effector and memory lymphocytes. The appropriate contact is composed of antigen complexed with MHC class II molecules on APCs, costimulators, and cytokines produced by the APCs and by the T cells themselves. This first encounter of naïve T cells with antigen is the primary immune response, which serves to induce the formation of effector and memory T cells. These activated T cells hone to peripheral tissues where, upon reexposure to their specific antigen, they activate macrophages to eliminate phagocytosed microbes and induce B-cell differentiation and antigen-specific antibody secretion. The CD8+ CTLs kill infected host cells and tumor cells that display class I MHC-associated antigen. Naïve T cells require activation by DCs, whereas effector T cells can respond to antigens presented by a wider variety of APCs, such as macrophages and B lymphocytes. Not surprisingly, differentiated effector and memory T cells possess lower thresholds for costimulation and require lower antigen concentration for activation than naïve T cells.
In general, antigen presented on MHC II molecules is the prototypical stimulus for CD4+ T-cell activation and the subsequent production of a variety of cytokine mediators, including IL-2, which stimulate further expansion and activation. However, the circumstances under which this activation occurs may dictate disparate paths of differentiation, producing T-cell subsets with distinct cytokine profiles and effector functions. These differing phenotypes have been utilized to characterize two distinct subsets: TH1 and TH2.59,60,74 IL-12 derived from phagocytes infected with intracellular pathogen provides the necessary signal for TH1 differentiation.39 IL-12 also stimulates production of IFNγ, the principle macrophage activator, by NK cells and CD4+ lymphocytes. Interferons stimulate TH1 development by augmenting phagocytic IL-12 production and by maintaining IL-12 receptor expression on CD4+ T cells. The principal effector action of TH1 cells is the activation of macrophages through the production of IFNγ, GM-CSF, TNFα, CD40L, and FasL.39,59,60,74 They regulate production of opsonizing and complement fixing antibodies and are effectors of phagocyte-dependent responses. This inflammatory response, also referred to as cell-mediated immunity or delayed-type hypersensitivity, provides one major arm of the adaptive immune response; it is mediated by CD4+ and CD8+ lymphocytes and macrophages.
The principal stimulus for TH2 differentiation is IL-4, which is derived from T cells, mast cells, and basophils.59,60,74 These cells are the cellular effectors of humoral immunity and provide the other major arm of the adaptive immune response, which is mediated by TH2 CD4+ cells, B cells, plasma cells, and antibodies. They produce IL-4, IL-5 and CD40L, thereby inducing B-cell activation and antibody production, and a host of other proinflammatory and anti-inflammatory cytokines.39 Activation of mast cells and eosinophils by extracellular pathogens is associated with activation of TH2 cells. TH2 cells quell the inflammatory response by inhibiting macrophage functions and TH1 responses. They are considered the anti-inflammatory arm of cell-mediated inflammation. Helper T cells that express both TH1 and TH2 patterns of cytokine expression have been called TH0 cells, and further studies will certainly discern other subsets of T cells.4,74
T-cell activation, differentiation, and expansion are orchestrated by the T cell itself. The responding T cell, in an autocrine fashion, serves as both source and target of a variety of mediators stimulating growth. The principal autocrine growth factor is IL-2, which is induced by signaling regulated by the phosphatase calcineurin (see below).75 IL-15 stimulates the proliferation of CD8+ T cells, especially memory cells of the CD8+ subset. After antigen exposure, the numbers of T cells specific for that antigen may increase to about 1 in 10 for CD8+ and 1 in 1000 to 10000 for CD4+ cells.4
After activation, some proliferating T cells will differentiate into effector cells that eliminate antigens and activate other immune cells. Mature CD4+ cells induce the activation of mononuclear phagocytes and B cells. CD8+ cells differentiate into CTL that recognize viral and other intracellular pathogen antigens that are presented in the context of MHC class I molecules and induce target cell death by releasing the cytotoxins perforin and granzymes from cytoplasmic granules. Granzymes are serine proteases that trigger DNA fragmentation and apoptosis. Perforin stimulates cell membrane pore formation that facilitates granzyme entrance into cells. Apoptosis can also be induced by the binding of Fas ligand on CTL to Fas on the target cell. CTLs also release the cytokines IFNγ, TNFα, and CC chemokines. IFNγ and certain CC chemokines have antiviral properties, and both are potent activators of macrophage function. IL-2 produced by CTL and local helper CD4+ lymphocytes expands the CTL, and IL-12 released by APC stimulates CTL activity. As with CD4+ cells, early evidence suggests that the population CD8+ may be divided into TC1 and TC2 cells based on their cytokine profiles and effector functions.39,58–60
Other T cells will mature into long-lived functionally quiescent memory cells. Upon antigen reexposure, a cell surface rich in adhesion molecules (i.e., integrins, CD44) facilitates rapid and efficient migration to peripheral sites of infection.4 These cells accumulate over time and in the adult human comprise more than half of the circulating T cells.
Mere antigen exposure is insufficient for activation of naïve T cells. Proliferation and differentiation require costimulatory signals provided by molecules on APCs. The best characterized costimulator pathway involves the T-cell surface molecule CD28 and its counterligands B7-1 and B7-2 expressed on activated APCs.4,39 CD28 delivers signals that enhance T-cell survival, by increasing expression of the antiapoptotic protein Bcl-X, the production of cytokines such as IL-2 and the IL-2 receptor, and the differentiation of immature T cells. In vitro, purified populations of CD4+ cells challenged with antigen by APCs that express B7, proliferate and secrete cytokines. This does not occur if B7 is absent. The costimulatory signal must come from the same APC that provides the initial signal. DCs are the most potent APC because they express both classes of MHC molecules and the B7 molecules, whereas macrophages and B cells must be activated to express the costimulatory molecules. This expression of costimulators is regulated so as to ensure that T-cell activation is temporally and spatially appropriate. For instance, during T-cell activation, engagement of CD40 ligand with CD40 induces upregulation of B7 costimulators on the APCs. In addition, it increases the secretion of cytokines such as IL-12 that promote T-cell differentiation, and cytokines are secreted that promote T-cell differentiation and activation. A protein CTLA-4, expressed on activated T cells, is homologous to CD28 and binds B7-1 and B7-2. Unlike CD28, CTLA-4 functions to terminate T-cell responses and plays a role in self-tolerance. On the basis of many experimental studies of costimulators, antagonists against B7 molecules and CD40L are in clinical trials to prevent the rejection of organ allografts.4,39
The differentiation of naïve CD8+ T cells to CTL requires a stronger costimulatory signal. This can be provided by either DC, as they have the greatest intrinsic costimulatory activity, or by a CD4+ helper T lymphocyte. Naïve helper T cells attached to the same APC as the CD8+ T cell can be activated to elaborate IL-2. Attached effector T helper cells can stimulate the APC to express more costimulatory molecules. In the case of virulent viruses, cytotoxicity can substitute for CD28 costimulation, and so the typical costimulatory signal is not required for activation. For less virulent viruses, costimulation is necessary for CTL induction.39,76 The absence of costimulation results in an unresponsive, or anergic T cell. Recently this has been shown to be mediated by the serine/threonine kinase calcium/calmodulin-dependent protein kinase (CaMK) II (see below).77 Anergic T cells do not produce IL-2 and therefore cannot proliferate and differentiate into effector cells even when presented with antigen at a later time.39
The affinity of most TCRs for peptide–MHC complexes is low, with dissociation constants of the order of 10−5 to 10−7 and an estimated TCR–antigen interaction of less than 10 seconds. Furthermore, on any APC fewer than 1000 of the 105 available MHC molecules are likely to be displaying any one peptide at any particular time. Therefore, one APC can engage a small fraction of the 104 to 105 antigen receptors on a single T cell.4 Activation of an individual T cell may require multiple sequential engagements of that cell’s antigen receptors by peptide–MHC complexes on APCs. With engagement, there is clustering of membrane receptors, tyrosine phosphorylation of several proteins, and recruitment and activation of adaptor proteins.
TCRs are devoid of enzymatic activity and must utilize other signal-transducing proteins.4 After TCR–MHC engagement, several membrane surface proteins and intracellular signaling proteins are rapidly recruited: TCR complex, CD4 or CD8, receptors for costimulators such as CD28, and enzymes and adaptor proteins.4 After TCR clustering, activated tyrosine kinases associated with and phosphorylate tyrosine residues on CD3 and TCR (Fig. 7-5). These phosphorylation sites provide docking sites for other tyrosine and protein kinases, such as Lck, an Src family tyrosine kinase, and ZAP-70, a tyrosine kinase. These kinases become activated with phosphorylation. Activated ZAP-70 phosphorylates several adaptor proteins that subsequently induce a variety of signal transduction cascades. Adaptor proteins contain structural domains that bind other proteins and thereby facilitate the correct spatial orientation that is required for signal transduction. The ras pathway is also activated, which is an early step in the activation of the mitogen-activated protein kinases (MAPK) that can activate a variety of transcription factors. Ras is a member of a family of guanine nucleotide-binding proteins (GDP/GTP) that are involved in diverse activation responses in different cell types. This pathway is an amplification process by which few upstream kinases lead to the activation of several downstream kinases. Ultimately activation of the terminal extracellular regulated kinase (ERK 1/2) leads to the phosphorylation of the protein ELK, which stimulates the transcription of fos, a component of the activation protein 1 (AP-1) transcription factor (Table 7-4). Concomitantly, c-Jun N-terminal kinase (JNK) is activated, which phosphorylates c-Jun, the second component of AP-1. The third member of the MAPK family, p38, is also activated. The activities of the MAPKs are terminated by specific protein tyrosine/threonine phosphatases that are regulated by the MAPKs themselves. Hence, the entire system is self-regulated by a negative feedback system.4

Figure 7-5. T-cell signaling and activation. (Redrawn from Abbas AK, Lichtman AH. Cellular and Molecular Immunology. 5th ed. Philadelphia, PA: Saunders; 2003.)
Table 7-4 Transcription Factors

Activation of TCR also leads to the induction of PLC, in particular PLCγ1 (Fig. 7-5). Phosphorylated PLCγ1 catalyzes the hydrolysis of phosphatidylinositol 4,5-bisphosphate (PIP2) into inositol 1,4,5-triphosphate (IP3) and diacylglycerol (DAG), and activates enzymes that generate additional active transcription factors. IP3 increases cytosolic calcium that leads to a large influx of both intracellular and extracellular calcium with subsequent activation of calcium- and calmodulin-dependent proteins. Calcineurin, a calcium/calmodulin-dependent phosphatase is integral to T-cell activation via modulation of the activation of the transcription factors nuclear factor of activated T cells (NFAT) and NFκB (Table 7-4).4,75 These transcription factors are essential for the induction of cytokine transcription, in particular IL-2 production. However, in the absence of costimulation, activation of the CaMK II opposes the actions of calcineurin as described above and produces an anergic cell.77 DAG activates protein kinase C, which activates additional transcription factors. The role of PKC and calcium in T-cell function is made evident by studies in which pharmacologic activation of PKC and/or elevation of intracellular calcium concentration stimulates T-cell cytokine secretion and proliferation.4 Regulation of those kinases operant in T-cell signaling involves protein tyrosine phosphatases. Through dephosphorylation they modulate TCR signaling. Two phosphatases induced with TCR clustering are SHP-1 and SHP-2.
The ultimate goal of all these signaling transduction pathways is to activate transcription factors that bind to promoter regions and enhance transcription. Three transcription factors that are activated in T cells and appear critical for most T-cell responses are NFAT, AP-1, and NFκB (Table 7-4).
A third mechanism of T-cell activation involves lipid antigens, such as cell wall protein from intracellular bacteria. These antigens bind CD1, a MHC-related cell surface molecule that presents these antigens to certain subtypes of T cells. A superantigen is an unprocessed bacterial or retroviral product that binds the MHC molecule and the TCR outside the usual antigen-binding sites. This engagement leads to a polyclonal and nonspecific stimulation of a large proportion of the T-cell population. An overwhelming activation of all arms of the immune system ensues and underlies much of the pathophysiology of toxic shock syndrome (TSS). Intravenous immunoglobulin (IVIG), by binding this antigen, is thought to be of therapeutic benefit.
Eosinophils
Eosinophils are marrow-derived granulocytes that share some properties of neutrophils, act in conjunction with basophils and mast cells as primary effectors in allergen inflammation and are involved in the eradication of helminthic infections (Table 7-1). Upon exiting the marrow, their intravascular half-life is but a few hours, whereafter they enter the mucosa of the lung, gastrointestinal, and genitourinary tracts.39,78 IL-3, GM-CSF, and IL-5 promote eosinophil differentiation, the induction of effector functions, and survival, the last by inhibiting apoptosis.78 Emigrating through inflamed endothelium they release inflammatory mediators and toxic agents from cytoplasmic granules. They generate superoxide anion and hydrogen peroxide, though less efficiently than neutrophils. They express IgE receptors and stimulate histamine release from basophils and mast cells through major basic protein (MBP). They can also regulate basophil and mast cell function by releasing enzymes that inactivate histamine and slow-reacting substance of anaphylaxis (SRS-A).
Recruitment and Activation
Eosinophils are recruited primarily to sites of parasitic infection and allergen challenge. Mast cells and macrophages responding to either secrete mediators (IL-5, PAF, LTB-4) that upregulate expression of endothelial adhesion molecules. Eosinophils themselves are more responsive to CC chemokines (MIP-1 α, RANTES, and MCP-3) and the cytokines produced with TH2 activation. Engagement of eosinophil β1 integrin VLA4 with VCAM-1 and fibronectin on the endothelium initiates the process of emigration. IL-4 can activate both the binding and the upregulation of endothelial cell VCAM-1. Diapedesis and tissue infiltration also involves members of the β2 integrin family (Mac-1 and LFA) similar to that which mediates neutrophil recruitment.39,79
Eosinophils are activated by IL-3, GM-CSF, IL-5, PAF, CC chemokines, and C3a and C5a of complement. IL-5 is a potent activating agent and enhances the ability of eosinophils to release granule contents on FcR cross-linking. A positive feedback cycle ultimately is established, in which recruited eosinophils produce cytokines and chemokines that recruit more eosinophils and other leukocytes.
Granules
Eosinophils possess a compartmentalized armamentarium of toxic substances that assist in the elimination of organisms, in particular helminths. Their specific granules contain GM-CSF and MBP, the latter of which is cytotoxic to parasites and normal cells. Further, it functions as a stimulus for histamine release from mast cells and basophils.80 The granule matrix contains eosinophil peroxidase (EPO), eosinophil-derived neurotoxin, lysosomal enzymes, catalase, TNFα, TGFβ, and eosinophilic cationic proteins that stimulate formation of transmembrane pores to increase target cellular permeability.81 EPO is released extracellularly on target cell surfaces where it generates hydrogen peroxide and hydrogen halides. Approximately 30% of oxygen consumed by stimulated eosinophils is utilized in the formation of halogenating species. Thiocyanate may be the major halide for the EPO–H2O2 system.82 If ingested by a neighboring phagocyte, EPO can combine with H2O2 and halides to form hypohalous acids. EPO also stimulates neutrophil aggregation and adhesion to endothelial cells. Although they express Fc receptors for IgG, IgA, and IgE, they are relatively insensitive to activation by antigen-mediated cross-linking of these receptors. However, they can kill microorganisms by antibody-dependent cell-mediated cytotoxicity.4,39
The primary targets of eosinophils are extracellular parasites. The size of these pathogens prohibits phagocytosis, and their integument is relatively resistant to the microbicidal products of neutrophils and macrophages; however they can be killed by MBP, which is released after cross-linking of Fc-bound IgE coating the parasite. The TH2 response to parasitic invasion produces IL-4, IL-5, and IL-13. IL-4 stimulates the production of specific IgE antibodies, which opsonize helminths. IL-5 activates eosinophils, which bind to the IgE-coated helminthes via Fc receptors. Activated eosinophils then release their granule contents and generate ROS and hypohalous acids. EPO also stimulates neutrophil aggregation and adhesion to endothelial cells. The sparse uncompartmental granules contain Charcot–Leyden crystals and have lysophospholipase activity. Finally, eosinophils produce and release lipid mediators such as PAF, prostaglandins, and LTs, which probably contribute to the process of allergic diseases.4,83,84
Basophils and Mast Cells
Basophils and mast cells are central to the development of allergic inflammation and produce cytokines, lipid mediators, vasoactive amines, and proteases that can also participate in nonallergic inflammatory responses (Table 7-1). Both cells share a common progenitor cell, which diverges early during differentiation. The primary growth factors for basophil development are IL-3 and GM-CSF, whereas stem cell factor is the primary stimulus for mast cell maturation.85 Although basophils and mast cells are distinct cell types, they are often grouped together because of the similarity of their granule content, the stimuli inducing activation, and their effector functions.39
Mature basophils constitute only 0.5% to 1% of the circulating leukocytes. Mast cells are primarily tissue-fixed residents of connective tissue. Engagement of IgE-bound antigen with the high-affinity IgE antibody receptor, FceR, provides the primary stimulus for basophil and mast cell activation and degranulation. In addition, diverse agents such as contrast media, opiates, anaphylatoxins, chemokines, and neuropeptides may serve as activators. Basophils and mast cells elaborate both preformed, as well as, newly synthesized inflammatory mediators. The main basophil proteoglycan is chondroitin sulfate A, whereas mast cells contain heparin, chondroitin sulfate, and chondroitin sulfate E and also store neutral proteases.86 The lipid mediators LTB4, LTC4, and PGD2 are synthesized de novo in response to stimulation. The LTs are powerful vasoconstrictors, bronchoconstrictors, and chemoattractants for neutrophils and eosinophils. PGD2 inhibits platelet aggregation and is chemotactic for neutrophils. Both basophils and mast cells synthesize and secrete histamine. Histamine, or 2-(4-imidazolyl)-ethylamine, is formed by the carboxylation of histidine. It is stored in preformed granules at acid pH as a complex with proteins and proteoglycans. Mast cells carry greater quantities of histamine than do basophils. The classic vasoactive properties of arteriolar dilatation, increased vascular permeability, and bronchoconstriction are mediated by the H1 receptors. H2 receptors are involved in modulation of the immune response and stimulating gastric output and mucus secretion. H3 receptors participate in neuroconduction.39,86,87
Platelets
Though classically considered in the context of hemostasis, studies confirm that platelets are also integral to normal and pathologic inflammation and link the processes of hemostasis, inflammation, and tissue repair. They are activated in a variety of inflammatory conditions, such as rheumatoid arthritis and inflammatory bowel disease, a testimony to their role in inflammation.88 Upon activation, they release factors that enhance vascular permeability, chemokines, microbicidal proteins, and mitogens for endothelial cells, smooth muscle cells, and fibroblasts. They assist leukocytes in promoting the inflammatory reaction and killing microbes by providing an adhesive surface to facilitate emigration, by stimulating adhered leukocytes, and by further modulating chemokine synthesis (Table 7-5).88,89
Humans possess about 150 to 400 × 109 platelets per liter of blood.88 Thrombopoiesis is regulated by thrombopoietin as well as a variety of other mediators (IL-3, IL-4, IL-6, IL-7, and IL-11). In fact, human IL-11 is in clinical use to stimulate thrombopoiesis in patients undergoing chemotherapy.90 After release from the marrow, platelets circulate with a half-life of approximately 12 days. Interferon α is inhibitory for megakaryocyte growth, and the elevated levels induced with some viral and inflammatory conditions may explain the relative thrombocytopenia observed in these conditions.88,91
Recruitment and Activation
Platelets are activated by and aggregate in response to thrombin or activated endothelium. Clot formation triggers the release of vasoactive agents (PGE2, PGI2, PGF2α, and platelet-derived growth factor [PDGF]) and inflammatory mediators. Serotonin, PGE2, and PAF increase vascular permeability. The adhesion molecule PECAM-1 and the β3 integrins mediate platelet plug formation, endothelial cell adhesion, and leukocyte emigration. The integrin IIb/IIIa and P-selectin facilitate platelet interactions with other inflammatory cells. The α granules are rich in membrane-bound P-selectin; upon fusion with the cell surface membrane they enhance the cell surface density of this receptor. On stimulation, both receptors are expressed on the platelet surface in an activated state. IIb/IIIa binds various adhesive proteins containing the RGD sequence (i.e., fibrinogen), which serve as a mechanical link to other platelets, leukocytes, and the endothelium. Possibly, the immobilization of activated P-selectin–expressing platelets on the vessel wall may biochemically and functionally promote the adhesion of neutrophils to endothelial cells.88,91
Table 7-5 Platelet-Derived Mediators

Platelets are a primary source of chemoattractants for neutrophils. Neutral proteinases released from stimulated platelets cleave complement factor C5, liberating the chemoattractant C5a. PDGF binds strongly to the extracellular matrix, providing a long-acting source of chemoattractant. Platelet factor 4 (PF4) is a cationic protein that penetrates the vascular wall and is a chemoattractant. Activated platelets also bind monocytes via P-selectin and PSGL-1 and induce the expression and secretion of monocyte chemotactic protein-1 (MCP-1) and IL-8. Thrombospondin released from activated platelets mediates monocyte binding to platelets. Together these substances promote leukocyte margination, activation, and recruitment to the sites of injury.39
Granules
The vasoactive substances and inflammatory mediators of platelets are either stored in cytoplasmic granules or synthesized de novo. A platelet contains about 35 α granules and 5 dense bodies. The dense granules contain adenosine diphosphate (ADP), ATP, serotonin, and calcium that are required during the earlier stages of inflammation. ADP is the principal platelet agonist during platelet aggregation and augments the oxidative burst of neutrophils. Serotonin increases vascular permeability and enhances the superoxide production by macrophages.88
The more abundant α granules contain fibrinogen, RANTES, MIP-1α, thrombospondin, P-selectin, PF4, PDGF, TGFβ, β-thromboglobulin, high–molecular-weight kininogen (HMWK), and many other biologically active proteins. PF4 and β-thromboglobulin initiate leukocyte recruitment and activation. PF4 induces neutrophil adherence to unstimulated endothelium and the release of secondary granules. It inhibits monocyte apoptosis and promotes macrophage differentiation. PF4 can also stimulate histamine release from basophils. RANTES is deposited on the endothelium and recruits monocytes from the circulation. PAF induces platelet aggregation, increases vascular permeability, enhances phagocyte free radical formation, and the adhesion of platelets to neutrophils. Platelet activation appears to occur in allergic asthma and may precede the delayed accumulation of eosinophils in the lung after allergen exposure. The α granules also carry several important growth factors, including vascular endothelial growth factor (VEGF), PDGF, and TGFβ. VEGF promotes extravasation and aids recruitment of leukocytes. PDGF is chemotactic for neutrophils and monocytes. TGFβ is chemotactic for and activates neutrophils and monocytes early during inflammation, but displays immunosuppressive effects during later stages of inflammation.88
Platelets are an important source of eicosanoids, including thromboxane, prostaglandins F2α and E2. Thromboxane synthetase in platelets is responsible for the production of TXA2, a potent vasoconstrictor that also increases vascular permeability and stimulates platelet aggregation. Aspirin and other nonsteroidal anti-inflammatory drugs (NSAIDS) inhibit platelet function by inhibiting TXA2 production. As this effect is irreversible, new platelets must be produced (7 to 10 days) to restore normal clot formation. In emergent circumstances (management of intracranial hemorrhage) platelet administration is required. Recently, an aspirin response test has been developed that assists in guiding therapy and targeting transfusion to those with functional evidence of platelet inhibition. It has also assisted in monitoring the response to transfusion. PGF2α causes vasoconstriction, whereas PGE2 vasodilates and modulates pain.92,93
Platelets participate in transcellular lipoxygenase (LO) metabolism, which refers to the production of eicosanoids through interactions with neighboring inflammatory cells. Endothelial cells utilize platelet-derived endoperoxides to synthesize PGI2. Platelets interact with neutrophils in several pathways, providing a direct link between thrombosis and inflammation. 12-hydroxyeicosatetraenoic acid (12-HETE) released from activated platelets can be used by unstimulated neutrophils to produce the chemoattractant 12,20-HETE. 12-HETE and 5-HETE from activated platelets and neutrophils, respectively, can combine in either cell type to form 5,12 diHETE, an anti-inflammatory compound, which diverts production away from the proinflammatory LTs. 12-LO from platelets and LTA4 formed by neutrophils can produce the intermediate 5(6)-epoxytetraene. This intermediate produces lipoxins A4 and B4 that have primarily counterinflammatory functions.39,89,92,94
In addition to modulating inflammation platelets possess some direct microbicidal activity. Platelets are activated and degranulate when exposed to certain bacteria. Electron microscopic studies have shown that activated platelets internalize bacteria and viruses. The α granules contain antibacterial proteins called thrombocidins (TC) in humans that support the killing of adherent bacteria. The two antibacterial proteins isolated from human platelets (TC-1, TC-2) are bactericidal for Escherichia coli and S. aureus.88
NONCELLULAR COMPONENTS
Cytokines
Cytokines are soluble protein mediators secreted by the cells of the innate and adaptive immunity in response to microbes and other antigens, including intra- and extracellular proteins, and mediate many of the functions of these cells (Table 7-6). They regulate and influence the host response to both PAMPs on bacterial, viruses, fungi, and parasites and DAMPs released during trauma, burns, allograft rejection, ischemia/reperfusion injury, and autoimmune disease. They govern lymphocyte differentiation during adaptive immunity and activate effector cells of both arms of inflammation to eliminate microbes. Cytokines play important roles in tumor biology and angiogenesis. Though essential for a normal immune response, excessive cytokine release underlies a variety of pathophysiological inflammatory states such as ARDS and MODS.95–98
Table 7-6 Cytokines

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


