“It has just stopped me cold from eating another burger!”
—Oprah Winfrey, during a segment on mad cow disease on her April 16, 1996, show
Prions and viroids are unconventional infectious agents. Prions are not viruses, bacteria, or parasites, but instead are molecules that cause fatal diseases in humans and animals. Prions can become attached to plants, such as grasses, or taken up by plant roots in contaminated soil. Prions are extremely stable in the environment for a long period of time and may be a source of transmission to animals eating prion-contaminated plants. Like prions, viroids are infectious molecules that only infect plants. Viroids are composed of RNA. Given their simple nature, prions and viroids find a home in virology textbooks.
Prions are proteins that cause a group of diseases of the brain and nervous system called transmissible spongiform encephalopathies (TSEs). TSEs affect humans, sheep, goats, mink, deer, elk, antelope, and cats (TABLES 18-1 and 18-2). None of the TSEs evoke a host immune response. Instead, they cause a noninflammatory process that results in the vacuolation, or spongiosis, in the gray matter of the brain. The observation that there is no inflammation or immune defense associated with prion diseases is fundamentally important for understanding the prion model of disease. Viroids are small pathogenic RNAs that cause virus-like diseases in plants. They do not code for proteins and depend on plant host enzymes for their replication and other functions.
Table 18-1 Human Prion Diseases
| Disease | Cause | Average Survival After Symptoms Appear |
|---|---|---|
| Kuru | Cannibalism | 3–6 months |
| Variant Creutzfeldt-Jakob disease (vCJD) | Consumption of contaminated beef (bovine prions) | 13–14 months |
| Creutzfeldt-Jakob disease | Inherited germline mutation in PrP gene, sporadic (unknown cause), or iatrogenic | 4–5 months |
| Gerstmann-Straussler-Scheinker disease (GSS) | Inherited germline mutation in PrP gene | 2–6 years |
| Fatal familial insomnia (FFI) | Inherited germline mutation in PrP gene | 12 months |
18.1 The “Mad” Diseases, Transmissible Spongiform Encephalopathies: Kuru and Cannibalism
Deadly Feasts by Richard Rhodes is a compelling account of prion diseases. The author begins the book by describing Kuru, a fatal brain disorder among the South Fore people who live in the highlands of Papua New Guinea. Kuru was at epidemic levels during the 1950s and 1960s. Kuru was known as “laughing sickness” because patients suffered from outbursts of laughter during the second stage of the disease. In the 1950s, scholars Vin Zigas, Shirley Lindenbaum, and Daniel Carleton Gadjusek traveled to New Guinea to study this mysterious disease. Gadjusek’s first impressions and observations of the Kuru epidemic were that the disease appeared to be “classical advancing Parkinsonism involving every age, overwhelming in females.” As fieldwork progressed, Lindenbaum reported that in the early 1960s fewer than 10% of females survived past child-bearing age, whereas males only had a 20% chance of dying of the disease. Gadjusek reported that Kuru victims suffered from three distinct stages of symptoms:
Ambulant stage: Patients suffered from an unsteady gait, voice, hands, and eyes; tremors and shivering; slurred and deteriorating speech; and loss of coordination in the lower extremities that slowly moved upward (FIGURE 18-1).
Sedentary stage: Patients could no longer walk without support; experienced tremors and coordination problems with increased severity; and suffered from jerky muscular movements, outbursts of laughter, depression, and mental slowing.
Terminal stage: Patients could not sit without support; tremors and slurring of speech continued in severity. Patients suffered from incontinence, difficulty swallowing, and deep ulcerations.
Table 18-2 Animal Prion Diseases
| Disease | Animal Host |
|---|---|
| Scrapie | Sheep and goats |
| Chronic wasting disease (CWD) | Deer and elk |
| Feline spongiform encephalopathy (FSE) | Cats |
| Bovine spongiform encephalopathy (BSE) | Cattle |
| Exotic ungulate encephalopathy (EUE) | Nyala and Greater Kudu (antelope in South Africa) |
| Transmissible mink encephalopathy (TME) | Mink |

FIGURE 18-1 A Fore child with symptoms of the ambulant stage of Kuru.
The Fore people of Papua New Guinea began eating their dead relatives (endocannibalism) during the early 1900s. The Fore ate their relatives as a sign of love and respect as part of their funeral rites. Everyone who died in the community was eaten. Human flesh was regarded as meat. The adult males preferred to eat domestic pigs or wild boars. Kuru victims were considered excellent sources of food because the fat of the victims resembled and tasted like pork.
Lindenbaum’s description (which can be found in Kuru Sorcery: Disease and Danger in the New Guinea Highlands, 1978) of this practice is as follows:
“After a member of the Fore tribe died, the mother of the next of kin would dismember the corpse. She removed the arms and feet and stripped the muscle from the limbs. Subsequently, the chest was cut open and the internal organs were removed. The head was severed from the skull and broken open. The brain was scooped out of the skull. Bones were broken and marrow was sucked out. The wife was expected to eat the penis of her dead husband. The entire body was eaten, even the feces. Care was taken not to rupture the gall-bladder because gallbladder contents seemed to spoil or ruin the taste of the body parts. A man’s brain was eaten by his sister. A woman’s brain was given to her son’s wife or her brother’s wife. Children under the age of ten and the elderly ate whatever the mothers’ ate and what was given to them: morsels of the brain or other internal organs. After the age of ten, the male children lived with the adult males. Males never ate the meat of females.”
Scientists came up with two main hypotheses to explain the cause of Kuru:
Kuru was a hereditary disease.
Endocannibalism was a practice associated with the transmission of an infectious agent that caused Kuru. People who ate their dead loved ones were infected with an infectious agent of unknown origin.
Heredity was considered unlikely rather quickly because the disease appeared recently in the Fore population (between 1900 and 1920). It quickly spread through a population of 40,000. Not all 40,000 Fore people could be descended from a single individual. During the early 1960s, Kuru reached epidemic numbers. About 1% of the South Fore people were dying of Kuru every year. Eight times more females than males contracted the disease. Females of all ages came down with Kuru; old men and young boys got the disease as well. It was determined that the practice of endocannibalism started 10 years before the first appearance of Kuru.
James McConnell authored a controversial paper regarding cannibalism in 1962. McConnell concluded that untrained flatworms (planaria) that were fed chopped-up trained flatworms retained some of the memories of the flatworms they ate. McConnell’s research pointed Kuru researchers to the idea that cannibalism can affect the brain and raised the question of whether Kuru and cannibalism were connected. Gajdusek’s team inoculated the brains of healthy chimpanzees with brain suspensions made from Kuru patients. The chimpanzees were held in isolation and observed for up to 5 years. Three chimpanzees developed Kuru-like symptoms similar to humans who suffered from Kuru. The average time it took for the chimpanzees to become sick was between 18 and 21 months—this is considered a very long incubation period. Their experiments led to the conclusion that a transmissible agent caused the Kuru-like disease, and that the agent was either a dormant or slow virus. In 1976, Gajdusek shared the Nobel Prize for Physiology or Medicine with Baruch S. Blumberg for their research on the origin and dissemination of infectious diseases. Blumberg gained notoriety for his work on the Australian antigen and the discovery of hepatitis B virus; Gajdusek received the award for his work on the origin of the class of diseases now called TSEs.
In 1972, Stanley Prusiner initiated research to isolate the infectious agent that caused Kuru and similar diseases such as Creutzfeldt-Jakob disease (CJD) and scrapie, a disease that affects sheep. Ten years later, Prusiner and his colleagues isolated a single infectious protein from diseased hamster brains that they named a “prion,” which stands for proteinaceous infectious particle (PrP). The scientific community was very skeptical about this research because the infectious agent did not contain any nucleic acid or hereditary DNA or RNA. In 1984, Prusiner’s team discovered that the gene encoding the prion was found in the brains of all the animals analyzed, including humans. It later became clear that there were two forms of the prion: the normal cellular protein PrPC and the other form of protein that caused diseases such as scrapies, termed PrPSC.
Another pioneer of prion research, Reed B. Wickner of the National Institutes of Health (NIH), reported in 1994 that a yeast prion called Ure2p could change shape and cause other yeast proteins to change to a similar shape. The Ure2p protein shared no sequence homology between the yeast and human protein. Wickner’s group continues to contribute to the field of prion research using yeast as a model. The research of Wickner’s team was central to the acceptance of Prusiner’s work. In 1997 Prusiner was awarded the Nobel Prize in Physiology or Medicine for his “discovery of prions—a new principal of infection.”
18.2 Characteristics and Formation of Infectious Prions
Prions are infectious proteins that are highly resistant to routinely used inactivation methods such as chemical decontamination, dry heat, or steam/autoclaving to sterilize surfaces. Prions are not inactivated by proteases, organic solvents, alkaline cleaners, ultraviolet light radiation, ethanol, formaldehyde, or extremely high temperatures. Even an extended sterilization run using an autoclave for 1 hour at 250°F (121°C) does not kill prions. Researchers working with tissues, infectious waste, and instruments used in the processing of prion-contaminated samples decontaminate them in 1 N NaOH (sodium hydroxide) or undiluted fresh household bleach followed by autoclaving at 270°F (132°C) for 4.5 hours. This procedure is much different from that of typical autoclave sterilization parameters to inactivate all other infectious agents. A standard autoclave run is 15 minutes at 15 pounds per square inch (psi) of steam at 249.8°F (121°C or 394.1°K).
A study led by Paul Brown at the National Institutes of Health (NIH) in Bethesda, Maryland, determined that incineration of prion-contaminated material for 15 minutes at 1,832°F (1,000°C) can destroy infectivity, but it is not a practical solution during a mass culling of large TSE outbreaks of bovine spongiform encephalopathy (BSE), chronic wasting disease (Cwd), or scrapie. Low infectivity remains after treatment at 1,112°F (600°C) for 15 minutes.
“Protein-Only” Hypothesis
The prion protein or PrP has two distinct conformations or isoforms: PrPC and PrPres. PrPC is the normal cellular form of PrP, and it is found throughout tissues of the body in healthy people and animals (FIGURE 18-2). The PrPC form is particularly abundant in neurons. PrPC is sensitive to denaturing or inactivation agents. The “protein-only” hypothesis proposes that the cellular form of the prion protein (PrPC) is posttranslationally misfolded into the infectious disease and resistant (to protease digestion) isoform denoted PrPres. Over time, the PrPres accumulates into clumps that damage or destroy nerve cells in the brain.
The PrPres isoform that causes scrapie in sheep is denoted as PrPSC (for scrapie prions). Purifying the infectious scrapie agent from hamster brains led to the amino acid sequence of PrPSC and, subsequently, to the identity of the encoding gene, PRNP. The PRNP gene is located on the short arm of chromosome 20 of humans and codes for PrPC. PrPC is a cell surface protein that is 254 amino acids in length (FIGURE 18-3). Sequence analysis indicates that the PRNP gene product is targeted by a secretory pathway to the cell surface of neurons and anchored into the plasma membrane by a glycosylphosphatidylinositol protein. While in the membrane, the prion may bind copper and is cycled back into the cell by endocytic vesicles, where it may be degraded in lysozymes (FIGURE 18-4). Experiments suggest that the infectious prions or PrPres isoform gain entry into endocytic vesicles where the PrPres isoform interacts with and changes the conformation of the wild-type noninfectious prion, PrPC, into the resistant form, PrPres. PrPres is resistant to protease degradation within cellular lysosomes. The infectious prions accumulate, causing neurotoxicity.

FIGURE 18-2 Tertiary structure of PrPC and PrPres proteins. The blue regions represent β sheets, the yellow regions are loops, and the green regions are α helices. The β sheets are thought to cause the amyloid clumping or aggregates in the brain.
Some researchers have hypothesized that other cellular factors stimulate the conversion of PrPC to PrPres. Investigators have demonstrated that vertebrate host ssRNAs are required for in vitro propagation of PrPres. It has been proposed that specific cellular ssRNAs bind avidly to and promote the conformational change of PrPC to PrPres.
Normal PrPC is highly conserved in mammals and predominantly found in the neurons of the brain. The exact function of PrPC is unknown. Substantial scientific evidence suggests that it is a copper-binding protein. In the conversion to the aberrant isoform of the protein, copper-binding ability is lost. Other studies suggest that PrPC binds other metals, such as manganese or zinc. The following have been postulated as possible functions for PrPC:

FIGURE 18-3 Biochemical analysis of the prion amino acid sequence. The prion contains a signal peptide that targets it to the surface of cells, a hydrophobic region that binds copper with a high affinity, and an octarepeat motif that contains amino acid residues that have low affinity for metals, especially copper. It is held together by a single disulfide bond.

FIGURE 18-4 Hypothetical model showing PrPC involved in the secretory pathway of cells. PrPres causes a conformational change in PrPC, resulting in the accumulation of prions resistant to lysosomal degradation.
Signal transduction
Cellular differentiation
Cell adhesion
Copper transport
Resistance to the accumulation of destructive free radicals that can result in neuronal death
The Three Ways That TSEs Can Arise: Infectious, Inherited, and Sporadic Forms
Sporadic CJD is the most common form of human TSE. Its cause is unknown, and patients usually die between the ages of 60 and 70 years. Other forms of TSEs are inherited. Examples of inherited forms are Creutzfeldt-Jakob disease (CJD), Gerstmann-Straussler-Scheinker disease, and fatal familial insomnia (FFI) (Table 18-1). In 5–15% of TSE cases, mutations or insertions of nucleotides in the PRNP gene are found. A history of TSE or dementia is recorded in less than 50% of cases.
Variant CJd is acquired through diet by eating beef contaminated with infectious prions. Kuru is a TSE acquired through endocannibalism. CJD can also be acquired by iatrogenic transmission, such as surgery, contaminated growth hormone injections, and corneal transplants.
18.3 Oral Transmission: How Do “Eaten” Prions Travel to the Brain to Cause Disease?
The transmission of TSEs such as variant CJD, Kuru, and BSE has been linked to the consumption of meat or meat-derived products contaminated with prions. The majority of experiments designed to study transmission of BSE and other TSEs involve animals of the same species or across different species using the intracranial route of transmission. It is known that oral transmission of TSE diseases is very ineff icient compared with intra-cerebral inoculations.
Neuroinvasion of the prions occurs by the splanchnic and vagus nerves present in the abdominal region of the body that extend to the brain. Research suggests that prions may also invade the brain through the hypoglossal nerve of the tongue. Bovine food products that contain tongue may be a potential source of prion infection for humans.
The infectious dose through ingestion of prion-contaminated food is unknown. A review of the risk assessment of exposure to BSE prions discharged into water from rendering plants, abattoirs (slaughterhouses), and landfills by Gale et al. (1998) estimated the human oral infectious dose, 50% (ID50) (the number of infectious agents needed to kill 50% of a population with that pathogen) to be 1013 BSE prion molecules. This is a very large infectious dose compared to known bacterial and viral pathogens. It was mistakenly believed that the minimum infectious dose for cattle was the ingestion of 1 gram of infected brain. The estimate caused concern about the potential risks of recycling BSE to cattle through the application of sewage sludge to agricultural land. A 4-mm mesh screen was placed over drains of slaughterhouses in the United Kingdom to prevent particles larger than 0.1 gram from entering the sewage system.
Some studies suggest that very low amounts of infectious prions ingested through diet may be enough to cause a subclinical infection in humans. The asymptomatic individuals would be carriers. The scale of the variant CJD epidemic was calculated to be a few thousand cases as an upper limit, but carriers might pose risk for contamination of blood products, surgical instruments, and tissue transplants. The variant CJD epidemic in the United Kingdom remains a public health concern and a subject of speculation.
18.4 Other Routes of Transmission: Iatrogenic Transmission, Including Prions in Blood
An iatrogenic disease is one that may be inadvertently caused by a physician or surgeon or by a contaminated medical or surgical instrument or diagnostic procedure. Iatrogenic transmission of CJD, which is rare, has been reported since the late 1970s. The following incidents have been associated with iatrogenic transmission of CJD:
Recipients of a corneal graft acquired CJD from donors who developed CJD.
Patients acquired CJD from contaminated depth electroencephalography (EEG) electrodes implanted into their brains.
Patients acquired CJD from contaminated neuro-surgical instruments.
Patients acquired CJD after acquiring human growth hormone from donors who developed CJD.
Patients acquired CJD after receiving dura mater grafts from donors who developed CJD.
The bloodborne transmission of variant CJD has long been suspected as being possible for two reasons:
The variant CJD agent can be detected in lymphoid tissues, raising the possibility that it could also be found in circulating lymphocytes present in blood.
The prions may exist in the blood as they travel from the original site of infection to the brain.
Experiments have demonstrated transmission of BSE to sheep by blood transfusion from asymptomatic sheep infected with BSE to healthy sheep, which means that the BSE agent is infectious in blood during the incubation period. Two highly probable bloodborne, person-to-person cases of transmission of variant CJD were reported in the United Kingdom. All cases of probable or definite variant CJD are reported to United Kingdom blood services by the national CJD surveillance unit. At least 48 individuals who received blood components from a total of 15 donors who later became variant CJD cases are being monitored as a precautionary step for their “at risk” status. Recipients of UK plasma products in the 1980s and early 1990s might also be at increased risk of variant CJD infection. For this reason, hemophiliacs in the United Kingdom have been notified that they are at an increased risk of variant CJD. TABLE 18-3 lists the precautionary steps introduced in the United Kingdom to reduce the risk of variant CJD transmission by blood and blood products. Surveillance systems record the actual chain of exposure to blood.
Table 18-3 Measures Taken in the United Kingdom to Reduce the Spread of Variant CJD Associated with Blood or Blood Products
| Year | Measure/Precautionary Step |
|---|---|
| 1997 | Blood components, plasma products, or tissues from an individual who contracted variant CJD were withdrawn or recalled. |
| 1998 | Plasma imported from the United States for fractionation (extracting and purifying proteins in plasma for patients). |
| 1998–1999 | Leukodepletion of all blood used for transfusion. |
| 2002 | Importation of fresh plasma from the United States for patients born after January 1, 1996. |
| 2004 | Blood donation is not accepted from people who have received a blood transfusion in the United Kingdom since 1980. |
| 2005 | Donors of blood to patients who have developed variant CJD following transfusion have been advised that they are “at risk” for variant CJD. |
18.5 Clinical Signs and Symptoms of Variant CJD
Over 50% of variant CJD patients die before the age of 30. The average age of death is 28 years. Patients suffering from sporadic or classic CJD die at an average of 68 years of age. The symptoms of variant CJD begin with anxiety, memory loss, mood changes, depression, and withdrawal, followed by obvious neurological signs that include muscle twitching or spasms (jerky movements) and posture and gait abnormalities (motor difficulties). Final symptoms are loss of speech, stupor, and a persistent vegetative state (coma) before death 14 months after symptoms appear.
18.6 Diagnosis of Variant CJD
Most patients with variant CJD are referred to a psychiatrist because of the behavioral changes associated with the disease. A definite diagnosis of variant CJD can be made by prion-positive immunostaining of biopsy material from the tonsils, spleen, or lymph nodes to detect prion accumulation. Other methods to aid diagnosis in conjunction with clinical symptoms are EEG (looking for slow or negative brain wave activity), magnetic resonance imaging (MRI, looking for brain lesions), and testing cerebral spinal fluid (CSF) for elevated levels of neuronal, astrocytic, and glial proteins. Elevated levels of CSF proteins reflect a change in blood–brain barrier function. The CSF proteins are released into the CSF as a consequence of extensive brain tissue damage.
There is no detectable immune response to infection, and as a result no routine test is available to diagnose asymptomatic individuals or patients experiencing symptoms. This rare but significant disease is difficult to diagnose with accuracy before death. The gold standard of diagnosis is the postmortem examination of brain tissues in order to detect abnormal protease-resistant PrPres by western blot analysis and immunocytochemistry of PrPres accumulation in brain tissues (FIGURE 18-5). Genetic studies may also be performed to examine whether the patient is homozygous for methionine at codon 129 of the prion protein.
18.7 Pathogenesis of TSEs
The incubation period of TSEs is long (20–56 years) with the exception of variant CJD. Individuals who lived in the South Fore in Papua New Guinea who were exposed during mortuary feasts are still dying of Kuru today, some 44 to 60 years after the cessation of endocannibalism.

FIGURE 18-5 (a) Detection of PrPres by western blot analysis. Brain homogenates are treated with proteinase K. Proteinase K will digest PrPC but not PrPres. Digests are electrophoretically separated on a polyacrylamide gel and then blotted onto a membrane that is probed with tagged anti-PrP antibodies. The arrows indicate PrPC or PrPres. The sporadic CJD brain contains a nonglycosylated PrPres protein (resulting in a double band). (b) Samples of human brain tissues from a diseased individual suspected of variant CJD.
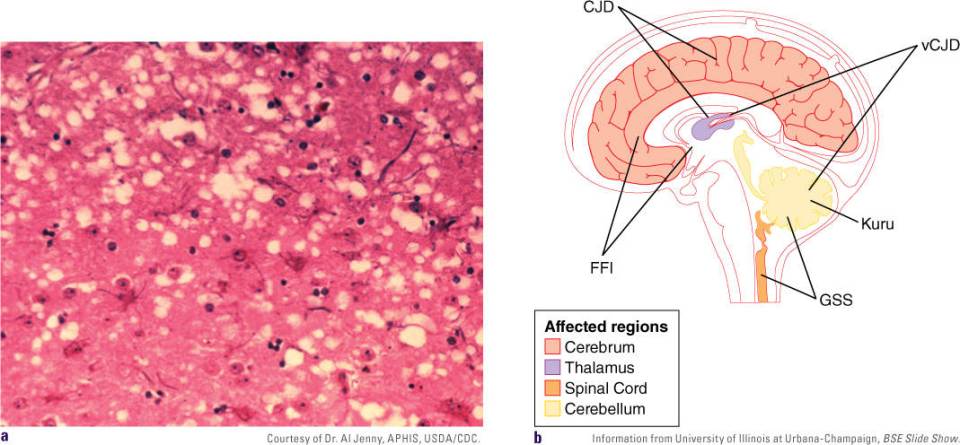
FIGURE 18-6 (a) Brain tissues showing histopathological changes found in bovine spongiform encephalopathy. Note the presence of vacuoles or spongiosis. (b) Diagram showing the major regions of the human brain affected by the different TSEs.
The brain, spinal cord, and retina are heavily infected before symptoms or clinical signs of infection appear. The term spongiform, which is sometimes referred to as spongiosis, comes from the microscopic observation of infected brains. Upon autopsy, slices of the brain contain vacuoles (clear zones) similar to a sponge (FIGURE 18-6A). Other classic histological changes of the brain are neuronal loss, astrocytosis (the spread of astrocytes to damaged tissues in the brain), and the formation of PrP thread-like structures that aggregate into amyloid plaques. The amyloid plaques accumulate in different regions of the brain (FIGURE 18-6B).
Depending on what region of the brain is affected, different symptoms that are typical for the particular type of disease are evident; for example, memory is affected when the cerebral cortex is infected. The precise mechanisms that lead to brain damage are not known; however, evidence suggests that apoptosis causes neuronal death. No inflammation or immune defenses against prions exist, presumably because they are natural proteins (i.e., the body does not recognize prions as being foreign). The prion proteins become harmful/infectious only when they are converted to PrPres.
18.8 The PRNP Gene
If the “protein-only” hypothesis holds true, then animals that lack the PRNP gene would be resistant to scrapie infection. Charles Weissmann’s team of researchers created knockout mice that did not express PrPC. A knockout mouse is a laboratory mouse in which scientists have inactivated, or “knocked out,” an existing gene by replacing it or disrupting it with noncoding DNA. When the PrPC knockout mice were injected with scrapie prions, the mice failed to develop symptoms of scrapie, an indication that PrPC is required for propagation of the infectious form of PrPres.
The idea that prions act as copper-binding proteins has been enticing to scientists. Some neurological disorders, such as Menkes syndrome and Wilson’s disease, are caused by altered copper metabolism. Could a change in copper binding be associated with the change to the pathogenic prion that is directly linked to TSEs? Weiss-mann’s group answered this question by introducing a PRNP gene that lacked the octarepeat copper-binding motif into the prion knockout mice (the gene that encodes the PrP protein was disrupted), and the mice became susceptible to infection by the prions that cause scrapie. This suggests that copper-binding octarepeat motif is not necessary for the pathogenic effects of prions. Does this mean that copper binding is not important in pathogenesis?
Researchers have found a different site on PrPC that is outside of the octarepeat region. It lies within the hydrophobic region of PrPC and binds copper strongly. The hydrophobic region is essential for disease propagation (Figure 18-3). Rather than PrPC copper binding being involved in transport, it may be involved in chelating free copper. Free copper radicals are toxic to cells and are usually quickly scavenged. Experiments in which radioactive copper was added to cells expressing PRNP in vitro revealed that the radioactive copper was bound to PrPC at the cell surface and then internalized. These cells were more resistant to copper toxicity and oxidative stress compared to cells that did not express PRNP. Hence, PrPC may play a role in cleaning up free radicals similar to other cellular scavenging mechanisms as a way to detoxify cells. The loss of PrPC function may change the copper balance in the brain and other organs and cause oxidative damage throughout the brain. Further research is needed to determine whether oxidative stress and copper activity are involved in the pathogenesis of TSEs.
Human Genetics: Codon 129 and Codon 219
At least 50 known mutations in the PRNP gene that result in spontaneous PRPres formation were identified in people who suffered from inherited TSEs such as classic CJD, fatal familial insomnia, or Gerstmann-StrausslerScheinker syndrome. A few of the mutations or common variations, referred to as polymorphisms in the PRNP gene, are recognized as increasing the susceptibility to TSEs (TABLE 18-4).
A common polymorphism at codon 129 of PRNP appears to act as a genetic susceptibility factor. Either methionine or valine is encoded at this position. All people suffering from variant CJD acquired through the consumption of prion-contaminated beef products have been homozygous for methionine at codon 129. Methionine homozygosity is generally associated with a rapidly progressive dementing disease, whereas a more prolonged course with an onset of ataxia (loss of motor coordination) is most often associated with the valine/valine genotype.
In contrast, heterozygosity of a codon 219 variant in which guanine is replaced by adenine in the first position of codon 219, resulting in a substitution from glutamic acid (E) to lysine (K), is considered protective or confers resistance against the sporadic form of CJD. The codon 219 variant is common in Japanese and Korean populations.
18.9 Steps Toward Treatment and Vaccination
The following compounds have been tested in cell lines and animal models: amphotericin B derivatives, Congo red, the spice curcumin, anthracyclines, polyanions, porphyrins, polyamines, β-sheet breakers, and even small interfering RNAs (siRNAs). Some promising in vitro prion therapy approaches have emerged, but none have been successful in human clinical trials due to toxicity or moderate untoward effects.
Table 18-4 Polymorphisms of the Human PRNP Gene
| Polymorphism | Disease Phenotype |
|---|---|
| Point mutation: 129 methionine or valine | Increases susceptibility to variant CJD but does not cause disease directly |
| Double point mutations: 171 asparagine or serine coupled and 129 methionine or valine | Increases susceptibility to variant CJD but does not cause disease directly |
| Point mutation: D178N (aspartic acid → asparagine) and 129 methionine | Fatal familial insomnia |
| Point mutation: V180I (valine → isoleucine) | Creutzfeldt-Jakob disease |
| Point mutation: T183A (threonine → alanine) | Creutzfeldt-Jakob disease |
| Point mutation: E200K (glutamic acid → lysine) | Creutzfeldt-Jakob disease |
| Point mutation: R208H (arginine to histidine) | Creutzfeldt-Jakob disease |
| Point mutation: V210I (valine → isoleucine) | Creutzfeldt-Jakob disease |
| Point mutation: M232R (methionine → arginine) | Creutzfeldt-Jakob disease |
| Point mutation: P102L (proline → leucine) and 129 methionine | Gerstmann-Straussler-Scheinker disease |
| Point mutation: P105L (proline → leucine) | Gerstmann-Straussler-Scheinker disease |
| Point mutation: A117V (alanine → valine) and 129 methionine | Gerstmann-Straussler-Scheinker disease |
| 48 base pair insertion | Creutzfeldt-Jakob disease |
| 96 base pair insertion | Creutzfeldt-Jakob disease |
| 120 base pair insertion | Creutzfeldt-Jakob disease |
| 192 base pair insertion | Gerstmann-Straussler-Scheinker disease |
| 216 base pair insertion | Gerstmann-Straussler-Scheinker disease |
Anti-PrPres antibodies can inhibit prion replication in cultured cells. Passive immunity experiments in which infected mice were injected with anti-PrPres anti-bodies produced a delay in the onset of symptoms. Any active vaccination experiments, however, have met with no success because the mammalian system is tolerant to PrPC. Studies in which antibodies against PrPC were injected into the brains of mice caused neurotoxicity. Vaccination efforts therefore should proceed through exhaustive safety trials in animals prior to application in humans.
18.10 Species Barrier: BSE and Variant CJD
Transmissibility of TSEs is easy among the same species. A high degree of homology in the amino acid sequence of the prion protein between two species increases the efficiency of transmission. It is also possible, though, for transmission to occur between different species by crossing the species barrier. The species barrier is a concept that explains why certain animal species are not infected by a given pathogen. The differences between species make it impossible for a pathogen to cross from one species to another. However, the species barrier is always under threat of mutation within that pathogen or other changes in a species that will affect its resistance against a pathogen, allowing an infectious agent to cross the species barrier.
History of BSE
The origin of BSE is unclear. It was hypothesized that it likely came from scrapie-infected sheep. The disease was spread to cattle by the ingestion of scrapie-contaminated bone meal derived from sheep offal fed to young calves. Offal consists of brain, spleen, thymus, tonsil, and guts— parts of the animal that are normally discarded. When bone meal of rendered bovine protein from infected cattle was fed to cattle to increase their milk yield, a new disease was further amplified among cattle.
Scrapie was considered a rare disease of sheep that did not infect humans. It was recognized as a disease of sheep and goats in Great Britain and other countries in Western Europe for more than 250 years. The first case of scrapie in the United States was diagnosed in 1947 in a flock of sheep in Michigan. The sheep owner had imported sheep of British origin through Canada for several years. Today, only Australia and New Zealand are free of scrapie.
BSE was first observed in 1985. A dairy farmer in England noticed several cows with abnormal behavior: unsteady gait, kicking unexpectedly during milking, and aggressiveness—hence the term “raging” or “mad cow” disease. The following are other signs of mad cow disease:
Difficulty in rising from a lying position
Itching
Heightened sensory perception
Anorexia
Excessive licking
Decreased milk production
The symptoms last for 2–6 months before the animal dies. In 1987, an official diagnosis of BSE was made after brain tissues of slaughtered, sick cows were similar to those of sheep suffering from scrapie. However, research suggests the first probable infections of BSE in cattle occurred during the 1980s, with two cases identified in 1986. The BSE epizootic in the United Kingdom peaked in January 1993, at almost 1,000 new cases per week. Over the next 17 years, the annual numbers of BSE dropped dramatically: 14,562 cases in 1995, 1,443 in 2000, 225 in 2005, and 11 in 2010. Cumulatively, through the end of 2005, more than 184,000 cattle in the United Kingdom were confirmed cases of mad cow disease and 1–3 million animals in more than 35,000 herds were likely to have been infected with the BSE agent, most of which were slaughtered for human consumption before developing signs of the disease. Cattle and feed exported from the United Kingdom seeded smaller epidemics in other European countries. In 1989, the United Kingdom implemented control measures that included the culling of all sick animals and the banning of all bovine materials from entering food that was fed to cattle.
In humans, the onset of variant CJD cases was first described in the United Kingdom from 1994 to 1996. The British Minister of Health declared that 10 people had died from a new form of CJD, a variant form. Transmission of variant CJD is believed to be caused by the ingestion of beef products contaminated with the BSE agent. The interval between the most likely period for the initial exposure of the population to potentially BSE-contaminated food (1984–1986) and the onset of initial variant CJD cases (1994–1996) is consistent with known incubation periods for the human forms of prion disease. As of June 1, 2015, 177 deaths of variant CJD patients had been reported in the United Kingdom since the start of the epidemic. The epidemic in cattle is largely under control, and any remaining risk to humans through the consumption of beef should be very small. Cases of BSE are diminishing in the United Kingdom, and only three cases of BSE in the United States have occurred. According to data collected between 1989 and 2015 by the Organization for Animal Health, 26 countries worldwide, excluding the United Kingdom, have reported BSE cases. The countries with the highest number of cases are France, Germany, Ireland, Portugal, Spain, and Switzerland.

FIGURE 18-7 BSE cases in North America, 1993 to February 18, 2015.
Through March 15, 2011, BSE surveillance has identified 22 cases in North America: 3 cases in the United States and 19 in Canada. The cases in the United States were cows imported from the United Kingdom into Canada or from Canada into the United States (FIGURE 18-7). Since March 2006, each of the 15 cattle reported with BSE in North America was born in Canada and identified through the Canadian BSE surveillance system. The Canadian Food Inspection Agency (CFIA) implemented an enhanced feed ban on July 12, 2007. On October 26, 2009, the U.S. Food and Drug Administration (FDA) followed by also issuing the enhanced feed ban. The enhanced BSE-related feed ban is a regulation or policy aimed to more effectively prevent and quickly eliminate BSE from Canada and the United States. Prior to the enhanced feed bans, the standard feed bans prohibited the feeding of most protein materials from mammals to ruminant animals. The enhanced feed ban prohibits most proteins, including the potentially BSE infectious tissues, not just from cattle feed but from all animal feeds, pet foods, and fertilizers. This practice is intended to eliminate any possibility of cross-contamination that was likely responsible for BSE in cattle born after the introduction of the feed ban.
Strains of BSE
The evidence increasingly suggests that there is more than one strain of BSE: the classic or typical BSE strain (C-type) and two atypical BSE strains (H- and L-types). The typical BSE strain is responsible for the outbreak in the United Kingdom and most of the BSE cases in Canada. It is preventable through the elimination of BSE-contaminated feed and was causally linked to variant CJD in humans. The typical strain has not yet been identified in the United States.
The atypical BSE strains were described in reports in 2004. Researchers assessed molecular and neuro-pathological (phenotypical) characteristics of seven cattle (three from France and four from Italy). Western blot analysis of the PrPres from brainstem tissues of sick cattle exhibited higher and lower molecular masses of unglycosylated PrPres. The types were named H-type and L-type. The L-type has now been identified in a number of different countries. In July 2007, the United Kingdom Spongiform Encephalopathy Advisory Committee announced that the atypical BSE strains may have risen spontaneously, but transmission through feed or the environment cannot be ruled out. Of the North American cases, three were linked to the H-type atypical strain of BSE and one was linked/identified as the L-type.
18.11 Chronic Wasting Disease
History
The deer population is being plagued by a new disease. Symptoms of the disease were first observed in mule deer grazing on northern Colorado wildlife research land in 1967. The deer showed chronic weight loss (ribs showing), blank facial expressions, excessive drooling and thirst, frequent urination, teeth grinding, holding head in a lowered position, nervousness, and sluggish behavior and were isolated from the herd. The sick animals had poor coats and appeared emaciated, or starving and “wasting away” (FIGURE 18-8). Most animals died within several months of illness onset, sometimes from aspiration pneumonia. Aspiration pneumonia is caused by breathing in foreign material such as foods, vomit, or liquids from the mouth into the lungs. It can lead to a lung abscess (collection of pus in the lungs), an inflammatory reaction, or a lung infection (pneumonia) that can result in death.

FIGURE 18-8 Deer suffering from CWD.
In 1978, researchers named the condition chronic wasting disease (CWD) and classified it as a TSE. In the mid-1980s, CWD was observed in free-range elk and deer in northern Colorado and adjacent parts of southern Wyoming. In 1997, it was observed in a farmed herd of elk in South Dakota. In 1999, the Wisconsin Department of Natural Resources (DNR) began to routinely test the state’s deer herd for CWD. In 2002, Wisconsin’s first cases of CWD were reported. The Wisconsin DNR continues to monitor and test for CWD during deer hunting season in specific locations in the state of Wisconsin. In 2013, the Wisconsin DNR reported that nearly 25% of the adult deer population in management zones where CWD was first detected tested positive for CWD. Between 2002 and 2011, more than $45 million was spent to manage CWD in Wisconsin. Much of the funding came from the federal government.
A map showing endemic locations of CWD in North America is shown in FIGURE 18-9A. Positive deer herds and farmed elk have been identified in 20 states at the time of this writing: Colorado, Illinois, Kansas, Maryland, Minnesota, Missouri, Montana, Nebraska, New Mexico, New York, North Dakota, Ohio, Oklahoma, Pennsylvania, South Dakota, Texas, Utah, West Virginia, Wisconsin, and Wyoming. During 2005 testing in Colorado, 1 out of 288 moose tested positive for CWD. It was not surprising that CWD is rarely found in moose given their social habits. Moose are typically solitary and stay with other moose only in cow–calf pairs. CWD was detected in wild deer and farmed elk in the Canadian provinces of Alberta and Saskatchewan. Outbreaks of CWD occurred in South Korea as a result of the importation of asymptomatic infected animals from Canada.
Animal-to-Animal Transmission
Whereas human prion diseases are very rare, CWD incidence can be over 15% in free-ranging deer and over 90% in captive deer herds. The incubation period of CWD is 1–5 years. Typically, symptoms are not seen in deer younger than 16 months old. Transmission of CWD prions (PrPCWD) between animals is much easier than that of BSE between cattle. Evidence suggests that infected deer or elk may transmit the CWD prions directly (animal-to-animal contact) or indirectly (through contaminated feed or water sources with saliva, urine, blood, feces, or decaying carcasses).
Prions are shed through the saliva, feces, blood, urine, or placenta of the infected animal. CWD is more likely to occur where deer or elk are crowded and congregate at artificial feeding, watering, and birthing sites. CWD is of particular concern because the PrPCWD is horizontally transmissible and remains infectious in the soil. A controlled lab study showed that the PrPCWD remained infectious for at least 2 years in a pasture.
It is likely that the environment serves as a stable reservoir of infectious CWD prions. A recent study evaluated whether plants could efficiently interact with prions to determine whether contaminated plants could play a role in natural prion transmission, especially in wild animals. It was demonstrated that wheat grass could uptake prions from contaminated soil and transport them to the stem and leaves of the plant. Hamsters fed prion-contaminated plant samples developed prion disease. These results suggest that plants can act as carriers of prions and play a role in horizontal transmission of prion diseases in the wild (FIGURE 18-9B). It is unclear if environmental transmission is sustained by mites, flies, predators, or scavengers (e.g., vultures, cougars). The stability of prions makes it difficult, if not impossible, to eliminate them from the environment.

FIGURE 18-9 (a) Map showing endemic locations of CWD in North America as of April 2016. (b) Prions present in the soil can be taken up by plant roots and transported to the stem and leaves of the plant. Prions in the environment can also bind and remain attached to plants for a long time. Animals in the wild can become infected if they eat the contaminated plants. Hamsters fed prion-contaminated plants develop prion diseases.
Diagnosis and Management
Hunting season typically occurs in the fall. The deer hunt plays an important role in wildlife management. Deer cause agricultural damage, deer–vehicle collisions, forest damage by overgrazing, damage to ornamental plants and landscaping, airport runway safety issues, and decreased food supply and cover for other species. CWD surveillance programs are now in place to identify CWD management zones. The gold standard for diagnosis of CWD is based on immunocytochemistry performed on tissues from the obex portion of the animal’s brain in a laboratory. A certified diagnostic test is not available for live animals. Managing CWD has become a major concern. The disease caused significant economic problems for game farmers after it was unintentionally introduced into farmed elk populations taken from CWD endemic areas.
Hunters’ Precautions
State fish and game departments are working hard to educate hunters, meat processors, taxidermists, and farmers. When field dressing carcasses, hunters in CWD endemic areas are asked to wear latex/rubber gloves and to carefully bone out the meat of the entire carcass. The catchphrase for returning home with deer and elk is “no skull, no backbone.” This is based on the idea that the prions are concentrated in the animal’s brain, spinal cord, and antlers. Removing them would reduce exposure to prions. Hunters are cautioned not to consume brains, spinal cords, eyes, spleens, tonsils, tongues, or lymph nodes of deer (FIGURE 18-10). Proper field dressing will remove most, if not all, of these body parts. A 2009 study by Angers and colleagues showed evidence that antler velvet from CWD-infected elk contained prions. Velvet is a highly innervated and vascularized skin layer that covers the growing antlers of male deer. The velvet is shed after the testosterone of the deer increases and the antlers become ossified. Antler velvet is sometimes used as a nutritional supplement for a variety of health remedies and health maintenance. Before this study, antler velvet was an unrecognized source of CWD prions in the environment.
Potential Transmission to Humans
CWD continues to spread among cervid populations (e.g., deer, elk, reindeer) in North America, creating concern that CWD may cross the species barrier to infect humans or domestic animals that may be consumed as food by humans. The most sensitive method to determine the susceptibility of TSE agents is by performing intra-cerebral injections of the PrPres of interest into a healthy host. Unfortunately, this type of experiment does not mimic most natural situations. Testing the susceptibility of infection of TSE agents through oral infection is used in species-barrier experiments to represent an oral route of transmission in laboratory investigations. In animal models excluding monkeys, oral transmission is generally 1,000-fold less effective than direct intracerebral injection challenge, resulting in longer incubation periods and lower efficiency of disease transmission.

FIGURE 18-10 Deer hunter Roger Gasser boning out the meat of a deer.
For example, in experiments in which ferrets were intracerebrally injected with PrPCWD, 100% were susceptible; however, the ferrets were not susceptible to oral infection. Mink were 25% susceptible to PrPCWD infection, but were not susceptible to oral infection.
CWD was successfully transmitted and adapted to laboratory hamsters, transgenic mice expressing hamster PrP, and transgenic mice overexpressing mouse PrP. However, transgenic mice overexpressing human PrP were not susceptible to CWD by intracerebral injection of PrPCWD. These experiments suggested a human species barrier against CWD infection.
Between January 1, 1997, and May 31, 2000, three young patients (a woman, age 28, and two men, ages 28 and 30) who regularly consumed venison were diagnosed with CJD. The cases were reported to the Centers of Disease Control and Prevention (CDC) out of concern about a possible zoonotic transmission of CWD. The cases were investigated, but no strong evidence supported a causal link between CWD and their illness. None of the patients had eaten deer or elk meat harvested in CWD-endemic areas of Colorado and Wyoming.
Research started by Richard F. Marsh in 1980 and published posthumously in 2005 demonstrated that two squirrel monkeys injected intracerebrally with brain homogenate from a mule deer suffering in the late stages of CWD developed a progressive TSE at 31 and 34 months following infection. The CWD-infected squirrel monkeys were euthanized during the terminal stages of disease. Their brain tissues were analyzed and found to contain PrPres. Histological examination of the brainstem and brain showed spongiform changes. This experiment provided evidence that at least one species of nonhuman primate was susceptible to CWD, reopening the possibility that humans may not be protected from CWD by a species barrier.
Race and colleagues followed Marsh’s work by carrying out further studies at the Rocky Mountain Laboratories (Hamilton, Montana). Their experiments involved cynomolgus macaques and squirrel monkeys (FIGURE 18-11). Humans are evolutionarily more closely related to cynomolgus macaques than to squirrel monkeys, making them a more accurate model for a human species barrier. In their study, 13 squirrel monkeys and 6 cynomolgus macaques were injected intracerebrally with CWD-positive brain homogenates. They also tested the more natural oral route of infection by giving 15 squirrel monkeys and 9 cynomolgus macaques oral doses of CWD-positive brain homogenates. The monkeys were anesthetized and given the oral doses through a rubber gastric tube. The brain homogenates were prepared from CWD-positive brains of free-ranging and captive mule deer, captive and wild white-tailed deer, and captive farmed elk located in Wyoming, Colorado, South Dakota, and Montana. The brain homogenates were pooled from eight different preparations of inoculum to make the CWD-positive brain homogenates.

FIGURE 18-11 Nonhuman primates used in experimental CWD studies. (a) Squirrel monkey. (b) Cynomolgus macaque.
A total of 11 out of 13 squirrel monkeys came down with a severe wasting syndrome similar to that of deer or elk within 33 to 53 months after the intracerebral injection. The monkeys were euthanized and their brains examined. The brains had spongiosis, and brain regions contained PrPres, as determined by immunocytochemistry. To date, three of the squirrel monkeys given oral doses of CWD brain homogenates came down with a severe wasting syndrome by 69 months postinoculation by oral dosage. Their brains had spongiosis and PrPres.
Eight years postinoculation, none of the cynomolgus macaques had signs of severe wasting disease through the intracerebral or oral route of transmission. Because cynomolgus macaques are evolutionarily closer to humans, this evidence suggests that humans may also be resistant to CWD. However, in experiments in which cynomolgus macaques were intracerebrally injected with BSE brain homogenates the human variant CJD disease occurred 3 years postinoculation and human sporadic CJD required 5 years. Oral inoculation of cynomolgus macaques with BSE brain homogenates took a minimum of 5 years before clinical disease was observed. Therefore, CWD transmission to cynomolgus macaques should not be ruled out.
Barria and colleagues published an in vitro study in 2011 that used a protein-misfolding cyclic amplification technique (PMCA). PMCA mimics protein replication in vitro at an accelerated speed. The misfolded form of PrPC made in vitro using PMCA is infectious in animals. New strains can be produced, adapted, and stabilized upon crossing species barriers in vitro by PMCA. In Barria’s study, the cervid PrPCWD triggered the conversion of human PrPC into PrPres, suggesting that CWD might be infectious to humans.
Human exposure to CWD-infected deer or elk meat in the past decades has likely occurred. The highest level of PrPres in cervids occurs in the CNS and lymphatic tissues of CWD-infected animals. The contamination of knives, saws, and muscles with these tissues can easily occur when field dressing game. Despite the high probability of hunters exposed, epidemiological studies of humans living in the CWD-endemic states of Colorado and Wyoming over 1979–2006 have not found any increases in human TSE cases.
The early signs of CWD in deer and elk were subtle behavior changes, staring, depression, and weight loss without a loss of appetite. The original explanation was that the wasting was caused by a metabolic disorder. It is possible that a wasting syndrome in humans may be diagnosed as a metabolic disorder as well, and the patient would never be tested for a TSE. Or is it possible that multiple infectious agents are involved? See VIRUS FILE 18-1. However, the epidemiological data, along with the in vivo cynomolgus macaque studies, are consistent with the conclusion that a species barrier protects humans from CWD infection.
18.12 Plant Viroids
Viroids are small RNA molecules that infect plants in the same manner as conventional plant viruses, but they have smaller genomes (246–401 nucleotides in length) and are not encapsidated in a protein coat. Viroid RNAs are positive-sense, single-stranded, covalently closed circular pathogenic molecules. Short helical regions are interrupted by internal and bulging loops. Base pairing often occurs across adjacent portions of the closed +ssRNA circular molecule, a process sometimes referred to as internal base-pairing. In nature, viroids assume a rodlike conformation (~37 nm) that is very stable against ribonucleases. Viroids do not code for any proteins, and some viroid RNAs even lack the AUG initiation codon. Viroid RNAs are classified as noncoding RNAs. Viroids depend upon the plant host enzymes for their replication and other functions.
The two families of viroids are the Avsunviroidae and the Pospiviroidae. Members of the Avsunviroidae family have a branched or “quasi” rodlike secondary structure, whereas members of the Pospiviroidae family possess a true rodlike secondary structure (FIGURE 18-12A–C). Viroids contain five structural or functional domains: central (C), pathogenicity (P), variable (V), and two terminal domains (T1 and T2; FIGURE 18-12d). Viroids of the Avsunviroidae family lack a C region but possess hammerhead ribozyme structures with self-cleaving properties. The C domain consists of conserved nucleotides in the upper and lower strands of the molecule. A secondary structure—the bulges—in the conserved C domain are critical for viroid replication and processing. The P domain is associated with symptoms of viroid-infected plants. The V domains contain the highest variability in nucleotide sequences between closely related viroids. The T domains are interchangeable between viroids and may play a role in viral movement.
Viroids do not replicate in the cytoplasm of cells like conventional plant RNA viruses. They instead traffic within the cell through the nuclear pores using a host nuclear localization protein that binds to viroid RNA. The viroids traffic from cell to cell through the plasmodesmata of the plant, traveling long distances through the phloem. All of these processes are associated with host proteins.
Viroids replicate in either the nucleus or the chloroplast of the plant. In the nucleus, the cellular DNA-dependent RNA polymerase III synthesizes the viroid RNA. The secondary structure of the viroid might provide binding signals to host factors directly or indirectly involved in the replication of the infectious agent. Members of the Avunsviroidae family accumulate and replicate in chloroplasts, whereas members of the Pospiviroidae family have a nuclear localization. It has been proposed that viroid replication occurs by the rolling circle model of replication. The host cell RNA polymerase synthesizes multiple copies of a linear complementary negative strand using the +ssRNA viroid genome as a template followed by the synthesis of the new, longer positive-strand RNA by the host cell RNA polymerase. The multiunit +ssRNAs undergo enzymatic self-cleavage by the hammerhead motifs of the viroid RNAs. A host RNA ligase may cause circularization of the plus strands (FIGURE 18-13). Viroid ssRNAs of positive polarity localize to the nucleolus and nucleoplasm of host cells. Viroid –ssRNA are localized only to the nucleoplasm.

FIGURE 18-12 Models of viroid structures. (a) Rodlike secondary structure of a member of the Pospiviroidae family. (b) “Quasi” rodlike secondary structure of the Avsunviroidae family member that has two terminal hairpins in the left part of the RNA molecule. (c) A member of the Avsunviroidae family with a complex branched structure. (d) Typical regions of a viroid: Terminal ends (T1 and T2), pathogenicity (P), conserved central region (C), and a variable domain (V).
More than 40 viroid species and variants have been characterized and cause at least 15 crop diseases (TABLE 18-5). “Classical” viroids were discovered only in plants. Human hepatitis delta virus RNA contains a viroid-like region with a secondary structure that has similar properties to viroid RNAs. Hepatitis delta virus also appears to replicate by a rolling circle mechanism. However, the RNA is larger (1.7 kilobases), is encapsidated, and encodes a virion-associated protein (hepatitis delta antigen). Viroids spread by mechanical transmission, vegetative propagation, and through pollen and seed. As a group, nothing distinguishes the disease symptoms produced by viroids from those caused by plant viruses. Both viroid and virus infections typically result in stunting, mottling, leaf distortion, and necrosis. Viroid diseases cause a range of symptoms, from a slowly developing lethal disease to more mild and asymptomatic infections. Viroids cause a number of serious diseases in economically important plants, including potato, tomato, coconut palm, grapevine, avocado, peach, apple, pear, citrus, coleus, and chrysanthemums.
Potato spindle tuber viroid (PSTVd) was the first viroid characterized. Theodor O. Diener, a plant pathologist at the U.S. Agricultural Research Service, discovered PSTVd in 1971. Tubers from diseased plants are long and narrow, with smooth skin and more eyes than are typically found in potatoes (FIGURE 18-14). The disease is spread easily by contact of healthy plants with diseased plants, by contaminated cultivating equipment, and through seeds and pollen. All potatoes and tomatoes are susceptible to PSTVd.

FIGURE 18-13 Rolling circle model (“toilet paper” model) of viroid RNA replication. The host cell RNA polymerase begins making multiple copies of the viroid genome, much like toilet paper being unraveled from a dispenser.
Table 18-5 Plant Diseases Caused by Viroids
| Viroid | Abbreviation |
|---|---|
| Apple dimple fruit viroid | ADFVd |
| Apple scar skin viroid | ASSVd |
| Australian grapevine viroid | AGVd |
| Avocado sunblotch viroid | ASBVd |
| Chrysanthemum stunt viroid | CSVd |
| Citrus bent leaf viroid | CBLVd |
| Citrus viroid species III | CVd-III |
| Citrus viroid species IV | CVd-IV |
| Citrus exocortis viroid | CEVd |
| Coconut cadang-cadang viroid | CCCVd |
| Coconut tinangaja viroid | CTiVd |
| Coleus blumei viroids I, II, and III | CBVd I, II and III |
| Columnea latent viroid | CLVd |
| Grapevine yellow speckle viroids 1 and 2 | GYSVd 1 and 2 |
| Hop latent viroid | HLVd |
| Hop stunt viroid | HSVd |
| Iresine viroid | IRVd |
| Mexican papita viroid | MPVd |
| Peach latent mosaic viroid | PLMVd |
| Pear blister canker viroid | PBCVd |
| Potato spindle tuber viroid | PSTVd |
| Tomato apical stunt viroid | TASVd |
| Tomato planta macho viroid | TPMVd |

FIGURE 18-14 Healthy tuber (top row) and diseased tubers caused by PSTVd (bottom row).

FIGURE 18-15 Chrysanthemums in the foreground of this photograph show symptoms of stunting and premature opening and color break caused by CSVd infection.
Chrysanthemum stunt disease crippled and nearly destroyed the chrysanthemum industry prior to 1950. It was prevalent in U.S. and Canadian greenhouses, with infection rates as high as 50–100%. Chrysanthemum stunt viroid (CSVd) is difficult to detect. It can be spread plant to plant by contaminated plant material, and it takes weeks for symptoms to appear after an infection. Symptoms of chrysanthemum stunt include shortened stems, uneven flowering (such as faded or irregular color), smaller than normal foliage, irregular flower sizes, and uneven maturation of parts within single flowers (FIGURE 18-15). Today chrysanthemums are grown in “clean” rooms using stringent precautions to prevent infection by CSVd.
Viroid Pathogenesis
The various effects of viroid infection are likely caused by more than one mechanism. Recent evidence suggests that pathogenic viroids activate a plant dsRnA-activated protein kinase (PKR). The plant PKR is analogous to the mammalian dsRNA-activated protein kinase PKR that is involved in setting up an antiviral state within the infected host cell through the induction of the interferon pathway.
Summary
Prions and viroids are pathogenic molecules. Both types of molecules are very stable in the environment. Prions are composed of protein, whereas viroids are closed, circular +ssRNAs that contain a lot of secondary structure. Neither code for proteins; both have self-replicating properties.
Prions cause transmissible spongiform encephalopathies (TSEs) in animal and human hosts. TSEs are rare, incurable, neurodegenerative fatal diseases. They do not induce an immune response. Creutzfeldt-Jakob disease (CJD) and variant CJD caused by eating prion-contaminated beef products are the most common TSEs in humans. The incubation period of TSEs is long (20–56 years). The brain, spinal cord, and retina are heavily infected before symptoms or clinical signs of infection appear. The term spongiform or spongiosis comes from the microscopic observation of infected brains of TSE patients or animals. Upon autopsy, slices of the brain contain vacuoles or clear zones, similar to a sponge. Other classic histological changes of the brain are neuronal loss, astrocytosis, and the formation of PrP threadlike structures that aggregate into amyloid plaques.
Prions are highly resistant to routinely used methods of decontamination and sterilization. Prions (PrP) have two distinct isoforms or conformations. The normal cellular isoform of PrP, designated PrPC, is sensitive to denaturing agents. Its exact function is unknown. The “protein-only” hypothesis proposes that the abnormal, misfolded infectious form of PrP is able to initiate a reaction that causes the PrPC to convert to the highly pathogenic and resistant, or stable, form termed PrPres. Over time the PrPres accumulates into clumps that damage or destroy neurons in the brain. At least 50 known mutations in the PRNP gene have been identified in people who suffer from inherited or sporadic TSEs such as classic CJD, fatal familial insomnia, or GerstmannStraussler-Scheinker syndrome.
Bovine spongiform encephalopathy (BSE), a disease in cattle, caused a mass epidemic in Great Britain, causing the death of more than 200,000 cattle during the 1980s to early 1990s. The BSE agent was shown to cause variant CJD in humans. Symptoms of TSEs in humans vary, but they commonly include personality changes, psychiatric problems such as depression, lack of coordination, and/or an unsteady gait. Patients may experience involuntary jerking movements called myoclonus, unusual sensations, insomnia, confusion, or memory problems. In the later stages of the disease, patients are severely mentally impaired and lose the ability to walk or speak. Death occurs within a few months to a few years. No treatment can halt the progression of TSEs.
Chronic wasting disease (CWD) is a contagious, fatal wasting disease of deer, elk, and moose caused by prions. Outbreaks have occurred in at least 20 U.S. states, in 2 Canadian provinces, and in imported cases in South Korea. The mode of transmission is not fully understood; however, evidence supports direct transmission through animal-to-animal contact and indirect transmission through exposure to the PrPCWD in the environment, including contaminated soil, plants, feed, and water sources. Surveillance of hunter-harvested deer and elk continues in order to identify endemic locations of the disease. Many humans consume deer and elk, raising cause for concern regarding transmission of CWD to humans. In vitro experiments using protein misfolding cyclic amplification (PMCA) implicate a possibility of CWD prions infecting humans. In vivo experiments challenging cynomolgus monkeys with CWD-positive brain homogenates and epidemiological studies suggest that a species barrier protects humans from CWD.
Viroids are small (about 300 nucleotides), infectious, nonencapsidated +ssRNAs that infect plants of economic importance. They reproduce by a process called the rolling circle model of replication. Viroids do not code for any proteins and are totally dependent upon and interact with host cell proteins for their replication and movement from cell to cell. Viroids have self-cleavage ribozyme activity. Nothing distinguishes plant infections caused by viroids from those caused by viruses. Despite their simplicity, viroids can cause varied symptoms, such as stunting, mottling, necrosis, and leaf distortion, or no symptoms at all. Viroids are difficult pathogens to control and can be catastrophic when large areas of food crops are destroyed by disease, threatening food supplies.

Full access? Get Clinical Tree


