FIGURE 103e-1 Gompertzian tumor growth. The growth fraction of a tumor declines exponentially over time (top). The growth rate of a tumor peaks before it is clinically detectable (middle). Tumor size increases slowly, goes through an exponential phase, and slows again as the tumor reaches the size at which limitation of nutrients or autoregulatory or host regulatory influences can occur. The maximum growth rate occurs at 1/e, the point at which the tumor is about 37% of its maximum size (marked with an X). Tumor becomes detectable at a burden of about 109 (1 cm3) cells and kills the patient at a tumor cell burden of about 1012 (1 kg). Efforts to treat the tumor and reduce its size can result in an increase in the growth fraction and an increase in growth rate.
LOCALIZED CANCER TREATMENTS
SURGERY
Surgery is unquestionably the most effective means of treating cancer. Today at least 40% of cancer patients are cured by surgery. Unfortunately, a large fraction of patients with solid tumors (perhaps 60%) have metastatic disease that is not accessible for removal. However, even when the disease is not curable by surgery alone, the removal of tumor can obtain important benefits, including local control of tumor, preservation of organ function, debulking that permits subsequent therapy to work better, and staging information on extent of involvement. Cancer surgery aiming for cure is usually planned to excise the tumor completely with an adequate margin of normal tissue (the margin varies with the tumor and the anatomy), touching the tumor as little as possible to prevent vascular and lymphatic spread, and minimizing operative risk. Such a resection is defined as an R0 resection. R1 and R2 resections, in contrast, are imprecisely defined pathologically as having microscopic or macroscopic tumor at resection margins. Such outcomes may be necessitated by proximity of the tumor to vital structures or recognition only in the resected specimen of the extent of tumor involvement, and may be the basis for reoperation to obtain optimal margins if feasible. Extending the procedure to resect draining lymph nodes obtains prognostic information and may, in some anatomic locations, improve survival.
Increasingly, laparoscopic approaches are being used to address primary abdominal and pelvic tumors. Lymph node spread may be assessed using the sentinel node approach, in which the first draining lymph node a spreading tumor would encounter is defined by injecting a dye or radioisotope into the tumor site at operation and then resecting the first node to turn blue or collect label. The sentinel node assessment is continuing to undergo clinical evaluation but appears to provide reliable information without the risks (lymphedema, lymphangiosarcoma) associated with resection of all the regional nodes. Advances in adjuvant chemotherapy (chemotherapy given systemically after removal of all disease by operation and without evidence of active metastatic disease) and radiation therapy following surgery have permitted a substantial decrease in the extent of primary surgery necessary to obtain the best outcomes. Thus, lumpectomy with radiation therapy is as effective as modified radical mastectomy for breast cancer, and limb-sparing surgery followed by adjuvant radiation therapy and chemotherapy has replaced radical primary surgical procedures involving amputation and disarticulation for childhood rhabdomyosarcomas and osteosarcomas. More limited surgery is also being used to spare organ function, as in larynx and bladder cancer. The magnitude of operations necessary to optimally control and cure cancer has also been diminished by technical advances; for example, the circular anastomotic stapler has allowed narrower (<2 cm) margins in colon cancer without compromise of local control rates, and many patients who would have had colostomies are able to maintain normal anatomy.
In some settings (e.g., bulky testicular cancer or stage III breast cancer), surgery is not the first treatment modality used. After an initial diagnostic biopsy, chemotherapy and/or radiation therapy is delivered to reduce the size of the tumor and clinically control undetected metastatic disease. Such therapy is followed by a surgical procedure to remove residual masses; this is called neoadjuvant therapy. Because the sequence of treatment is critical to success and is different from the standard surgery-first approach, coordination among the surgical oncologist, radiation oncologist, and medical oncologist is crucial.
Surgery may be curative in a subset of patients with metastatic disease. Patients with lung metastases from osteosarcoma may be cured by resection of the lung lesions. In patients with colon cancer who have fewer than five liver metastases restricted to one lobe and no extrahepatic metastases, hepatic lobectomy may produce long-term disease-free survival in 25% of selected patients. Surgery can also be associated with systemic antitumor effects. In the setting of hormonally responsive tumors, oophorectomy and/or adrenalectomy may eliminate estrogen production, and orchiectomy may reduce androgen production, hormones that drive certain breast and all prostate cancers, respectively; both procedures can have useful effects on metastatic tumor growth. If resection of the primary lesion takes place in the presence of metastases, acceleration of metastatic growth has also been described in certain cases, perhaps based on the removal of a source of angiogenesis inhibitors and mass-related growth regulators in the tumor.
In selecting a surgeon or center for primary cancer treatment, consideration must be given to the volume of cancer surgeries undertaken by the site. Studies in a variety of cancers have shown that increased annual procedure volume appears to correlate with outcome. In addition, facilities with extensive support systems—e.g., for joint thoracic and abdominal surgical teams with cardiopulmonary bypass, if needed—may allow resection of certain tumors that would otherwise not be possible.
Surgery is used in a number of ways for palliative or supportive care of the cancer patient, not related to the goal of curing the cancer. These include insertion and care of central venous catheters, control of pleural and pericardial effusions and ascites, caval interruption for recurrent pulmonary emboli, stabilization of cancer-weakened weight-bearing bones, and control of hemorrhage, among others. Surgical bypass of gastrointestinal, urinary tract, or biliary tree obstruction can alleviate symptoms and prolong survival. Surgical procedures may provide relief of otherwise intractable pain or reverse neurologic dysfunction (cord decompression). Splenectomy may relieve symptoms and reverse hypersplenism. Intrathecal or intrahepatic therapy relies on surgical placement of appropriate infusion portals. Surgery may correct other treatment-related toxicities such as adhesions or strictures. Surgical procedures are also valuable in rehabilitative efforts to restore health or function. Orthopedic procedures may be necessary to ensure proper ambulation. Breast reconstruction can make an enormous impact on the patient’s perception of successful therapy. Plastic and reconstructive surgery can correct the effects of disfiguring primary treatment.
Surgery is also a tool valuable in the prevention of cancers in high-risk populations. Prophylactic mastectomy, colectomy, oophorectomy, and thyroidectomy are mainstays of prevention of genetic cancer syndromes. Resection of premalignant skin and uterine cervix lesions and colonic polyps prevents progression to frank malignancy.
RADIATION
Radiation Biology and Medicine Therapeutic radiation is ionizing; it damages any tissue in its path. The selectivity of radiation for causing cancer cell death may be due to defects in a cancer cell’s ability to repair sublethal DNA and other damage. Ionizing radiation causes breaks in DNA and generates free radicals from cell water that may damage cell membranes, proteins, and organelles. Radiation damage is augmented by oxygen; hypoxic cells are more resistant. Augmentation of oxygen presence is one basis for radiation sensitization. Sulfhydryl compounds interfere with free radical generation and may act as radiation protectors. X-rays and gamma rays are the forms of ionizing radiation most commonly used to treat cancer. They are both electromagnetic, nonparticulate waves that cause the ejection of an orbital electron when absorbed. This orbital electron ejection is called ionization. X-rays are generated by linear accelerators; gamma rays are generated from decay of atomic nuclei in radioisotopes such as cobalt and radium. These waves behave biologically as packets of energy, called photons. Particulate ionizing radiation using protons has also become available. Most radiation-induced cell damage is due to the formation of hydroxyl radicals from tissue water:
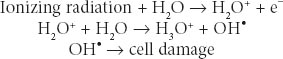
Radiation is quantitated based on the amount of radiation absorbed by the tumor in the patient; it is not based on the amount of radiation generated by the machine. The International System (SI) unit for radiation absorbed is the Gray (Gy): 1 Gy refers to 1 J/kg of tissue; 1 Gy equals 100 centigrays (cGy) of absorbed dose. A historically used unit appearing in the oncology literature, the rad (radiation absorbed dose), is defined as 100 ergs of energy absorbed per gram of tissue and is equivalent to 1 cGy. Radiation dosage is defined by the energy absorbed per mass of tissue. Radiation dose is measured by placing detectors at the body surface or based on radiating phantoms that resemble human form and substance, containing internal detectors. The features that make a particular cell more sensitive or more resistant to the biologic effects of radiation are not completely defined and critically involve DNA repair proteins that, in their physiologic role, protect against environmentally related DNA damage.
Localized Radiation Therapy Radiation effect is influenced by three determinants: total absorbed dose, number of fractions, and time of treatment. A frequent error is to omit the number of fractions and the duration of treatment. This is analogous to saying that a runner completed a race in 20 s; without knowing how far he or she ran, the result is difficult to interpret. The time could be very good for a 200-m race or very poor for a 100-m race. Thus, a typical course of radiation therapy should be described as 4500 cGy delivered to a particular target (e.g., mediastinum) over 5 weeks in 180-cGy fractions. Most curative radiation treatment programs are delivered once a day, 5 days a week, in 150- to 200-cGy fractions.
A number of parameters influence the damage done to tissue (normal and tumor) by radiation. Hypoxic cells are relatively resistant. Nondividing cells are more resistant than dividing cells, and this is one rationale for delivering radiation in repeated fractions, to ultimately expose a larger number of tumor cells that have entered the division cycle. In addition to these biologic parameters, physical parameters of the radiation are also crucial. The energy of the radiation determines its ability to penetrate tissue. Low-energy orthovoltage beams (150–400 kV) scatter when they strike the body, much like light diffuses when it strikes particles in the air. Such beams result in more damage to adjacent normal tissues and less radiation delivered to the tumor. Megavoltage radiation (>1 MeV) has very low lateral scatter; this produces a skin-sparing effect, more homogeneous distribution of the radiation energy, and greater deposit of the energy in the tumor, or target volume. The tissues that the beam passes through to get to the tumor are called the transit volume. The maximum dose in the target volume is often the cause of complications to tissues in the transit volume, and the minimum dose in the target volume influences the likelihood of tumor recurrence. Dose homogeneity in the target volume is the goal. Computational approaches and delivery of many beams to converge on a target lesion are the basis for “gamma knife” and related approaches to deliver high doses to small volumes of tumor, sparing normal tissue.
Therapeutic radiation is delivered in three ways: (1) teletherapy, with focused beams of radiation generated at a distance and aimed at the tumor within the patient; (2) brachytherapy, with encapsulated sources of radiation implanted directly into or adjacent to tumor tissues; and (3) systemic therapy, with radionuclides administered, for example, intravenously but targeted by some means to a tumor site. Teletherapy with x-ray or gamma-ray photons is the most commonly used form of radiation therapy. Particulate forms of radiation are also used in certain circumstances, such as the use of proton beams. The difference between photons and protons relates to the volume in which the greatest delivery of energy occurs. Typically protons have a much narrower range of energy deposition, theoretically resulting in more precise delivery of radiation with improvement in the degree to which adjacent structures may be affected, in comparison to photons. Electron beams are a particulate form of radiation that, in contrast to photons and protons, have a very low tissue penetrance and are used to treat cutaneous tumors. Apart from sparing adjacent structures, particulate forms of radiation are in most applications not superior to x-rays or gamma rays in clinical studies reported thus far, but this is an active area of investigation.
Certain drugs used in cancer treatment may also act as radiation sensitizers. For example, compounds that incorporate into DNA and alter its stereochemistry (e.g., halogenated pyrimidines, cisplatin) augment radiation effects at local sites, as does hydroxyurea, another DNA synthesis inhibitor. These are important adjuncts to the local treatment of certain tumors, such as squamous head and neck, uterine cervix, and rectal cancers.
Toxicity of Radiation Therapy Although radiation therapy is most often administered to a local region, systemic effects, including fatigue, anorexia, nausea, and vomiting, may develop that are related in part to the volume of tissue irradiated, dose fractionation, radiation fields, and individual susceptibility. Injured tissues release cytokines that act systemically to produce these effects. Bone is among the most radioresistant organs, with radiation effects being manifested mainly in children through premature fusion of the epiphyseal growth plate. By contrast, the male testis, female ovary, and bone marrow are the most sensitive organs. Any bone marrow in a radiation field will be eradicated by therapeutic irradiation. Organs with less need for cell renewal, such as heart, skeletal muscle, and nerves, are more resistant to radiation effects. In radiation-resistant organs, the vascular endothelium is the most sensitive component. Organs with more self-renewal as a part of normal homeostasis, such as the hematopoietic system and mucosal lining of the intestinal tract, are more sensitive. Acute toxicities include mucositis, skin erythema (ulceration in severe cases), and bone marrow toxicity. Often these can be alleviated by interruption of treatment.
Chronic toxicities are more serious. Radiation of the head and neck region often produces thyroid failure. Cataracts and retinal damage can lead to blindness. Salivary glands stop making saliva, which leads to dental caries and poor dentition. Taste and smell can be affected. Mediastinal irradiation leads to a threefold increased risk of fatal myocardial infarction. Other late vascular effects include chronic constrictive pericarditis, lung fibrosis, viscus stricture, spinal cord transection, and radiation enteritis. A serious late toxicity is the development of second solid tumors in or adjacent to the radiation fields. Such tumors can develop in any organ or tissue and occur at a rate of about 1% per year beginning in the second decade after treatment. Some organs vary in susceptibility to radiation carcinogenesis. A woman who receives mantle field radiation therapy for Hodgkin’s disease at age 25 years has a 30% risk of developing breast cancer by age 55 years. This is comparable in magnitude to genetic breast cancer syndromes. Women treated after age 30 years have little or no increased risk of breast cancer. No data suggest that a threshold dose of therapeutic radiation exists below which the incidence of second cancers is decreased. High rates of second tumors occur in people who receive as little as 1000 cGy.
OTHER LOCALIZED CANCER TREATMENTS
Endoscopy techniques may allow the placement of stents to unblock viscera by mechanical means, palliating, for example, gastrointestinal or biliary obstructions. Radiofrequency ablation (RFA) refers to the use of focused microwave radiation to induce thermal injury within a volume of tissue. RFA can be useful in the control of metastatic lesions, particularly in liver, that may threaten biliary drainage (as one example) and threaten quality and duration of useful life in patients with otherwise unresectable disease. Cryosurgery uses extreme cold to sterilize lesions in certain sites, such as prostate and kidney, when at a very early stage, eliminating the need for modalities with more side effects such as surgery or radiation.
Some chemicals (porphyrins, phthalocyanines) are preferentially taken up by cancer cells by mechanisms not fully defined. When light, usually delivered by a laser, is shone on cells containing these compounds, free radicals are generated and the cells die. Hematoporphyrins and light (phototherapy) are being used with increasing frequency to treat skin cancer; ovarian cancer; and cancers of the lung, colon, rectum, and esophagus. Palliation of recurrent locally advanced disease can sometimes be dramatic and last many months.
Infusion of chemotherapeutic or biologic agents or radiation-bearing delivery devices such as isotope-coated glass spheres into local sites through catheters inserted into specific vascular sites such as liver or an extremity have been used in an effort to control disease limited to that site; in selected cases, prolonged control of truly localized disease has been possible.
SYSTEMIC CANCER TREATMENTS
The concept that systemically administered agents may have a useful effect on cancers was historically derived from three sets of observations. Paul Ehrlich in the nineteenth century observed that different dyes reacted with different cell and tissue components. He hypothesized the existence of compounds that would be “magic bullets” that might bind to tumors, owing to the affinity of the agent for the tumor. A second observation was the toxic effects of certain mustard gas derivatives on the bone marrow during World War I, leading to the idea that smaller doses of these agents might be used to treat tumors of marrow-derived cells. Finally, the observation that certain tumors from hormone-responsive tissues, e.g., breast tumors, could shrink after oophorectomy led to the idea that endogenous substances promoting the growth of a tumor might be antagonized. Chemicals achieving each of the goals are actually or intellectually the forbearers of the currently used cancer chemotherapy agents.
Systemic cancer treatments are of four broad types. Conventional “cytotoxic” chemotherapy agents were historically derived by the empirical observation that these “small molecules” (generally with molecular mass <1500 Da) could cause major regression of experimental tumors growing in animals. These agents mainly target DNA structure or segregation of DNA as chromosomes in mitosis. Targeted agents refer to small molecules or “biologics” (generally macromolecules such as antibodies or cytokines) designed and developed to interact with a defined molecular target important in maintaining the malignant state or expressed by the tumor cells. As described in Chap. 102e, successful tumors have activated biochemical pathways that lead to uncontrolled proliferation through the action of, e.g., oncogene products, loss of cell cycle inhibitors, or loss of cell death regulation, and have acquired the capacity to replicate chromosomes indefinitely, invade, metastasize, and evade the immune system. Targeted therapies seek to capitalize on the biology behind the aberrant cellular behavior as a basis for therapeutic effects. Hormonal therapies (the first form of targeted therapy) capitalize on the biochemical pathways underlying estrogen and androgen function and action as a therapeutic basis for approaching patients with tumors of breast, prostate, uterus, and ovarian origin. Biologic therapies are often macromolecules that have a particular target (e.g., antigrowth factor or cytokine antibodies) or may have the capacity to regulate growth of tumor cells or induce a host immune response to kill tumor cells. Thus, biologic therapies include not only antibodies but also cytokines and gene therapies.
CANCER CHEMOTHERAPY
Principles The usefulness of any drug is governed by the extent to which a given dose causes a useful result (therapeutic effect; in the case of anticancer agents, toxicity to tumor cells) as opposed to a toxic effect to the host. The therapeutic index is the degree of separation between toxic and therapeutic doses. Really useful drugs have large therapeutic indices, and this usually occurs when the drug target is expressed in the disease-causing compartment as opposed to the normal compartment. Classically, selective toxicity of an agent for a tissue or cell type is governed by the differential expression of a drug’s target in the “sensitive” cell type or by differential drug accumulation into or elimination from compartments where greater or lesser toxicity is experienced, respectively. Currently used chemotherapeutic agents have the unfortunate property that their targets are present in both normal and tumor tissues. Therefore, they have relatively narrow therapeutic indices.
Figure 103e-2 illustrates steps in cancer drug development. Following demonstration of antitumor activity in animal models, potentially useful anticancer agents are further evaluated to define an optimal schedule of administration and arrive at a drug formulation designed for a given route of administration and schedule. Safety testing in two species on an analogous schedule of administration defines the starting dose for a phase 1 trial in humans, usually but not always in patients with cancer who have exhausted “standard” (already approved) treatments. The initial dose is usually one-sixth to one-tenth of the dose just causing easily reversible toxicity in the more sensitive animal species. Escalating doses of the drug are then given during the human phase 1 trial until reversible toxicity is observed. Dose-limiting toxicity (DLT) defines a dose that conveys greater toxicity than would be acceptable in routine practice, allowing definition of a lower maximum-tolerated dose (MTD). The occurrence of toxicity is, if possible, correlated with plasma drug concentrations. The MTD or a dose just lower than the MTD is usually the dose suitable for phase 2 trials, where a fixed dose is administered to a relatively homogeneous set of patients with a particular tumor type in an effort to define whether the drug causes regression of tumors. In a phase 3 trial, evidence of improved overall survival or improvement in the time to progression of disease on the part of the new drug is sought in comparison to an appropriate control population, which is usually receiving an acceptable “standard of care” approach. A favorable outcome of a phase 3 trial is the basis for application to a regulatory agency for approval of the new agent for commercial marketing as safe and possessing a measure of clinical effectiveness.
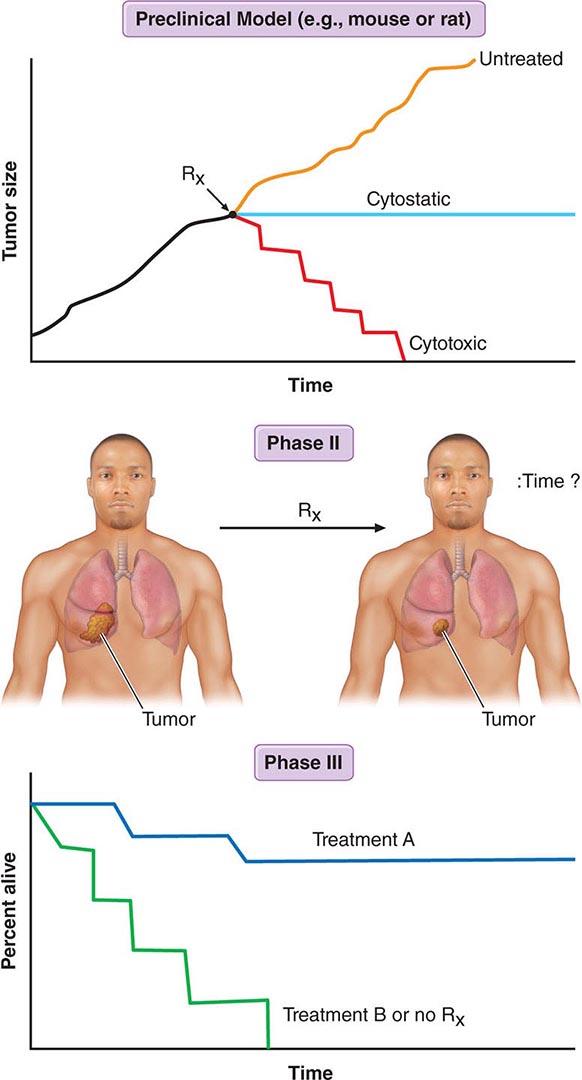
FIGURE 103e-2 Steps in cancer drug discovery and development. Preclinical activity (top) in animal models of cancers may be used as evidence to support the entry of the drug candidate into phase 1 trials in humans to define a correct dose and observe any clinical antitumor effect that may occur. The drug may then be advanced to phase 2 trials directed against specific cancer types, with rigorous quantitation of antitumor effects (middle). Phase 3 trials then may reveal activity superior to standard or no treatment (bottom).
Response, defined as tumor shrinkage, is the most immediate indicator of drug effect. To be clinically valuable, responses must translate into clinical benefit. This is conventionally established by a beneficial effect on overall survival, or at least an increased time to further progression of disease. Karnofsky was among the first to champion the evaluation of a chemotherapeutic agent’s benefit by carefully quantitating its effect on tumor size and using these measurements to objectively decide the basis for further treatment of a particular patient or further clinical evaluation of a drug’s potential. A partial response (PR) is defined conventionally as a decrease by at least 50% in a tumor’s bidimensional area; a complete response (CR) connotes disappearance of all tumor; progression of disease signifies an increase in size of existing lesions by >25% from baseline or best response or development of new lesions; and stable disease fits into none of the above categories. Newer evaluation systems, such as Response Evaluation Criteria in Solid Tumors (RECIST), use unidimensional measurement, but the intent is similar in rigorously defining evidence for the activity of the agent in assessing its value to the patient. An active chemotherapy agent conventionally has PR rates of at least 20–25% with reversible non-life-threatening side effects, and it may then be suitable for study in phase 3 trials to assess efficacy in comparison to standard or no therapy. Active efforts are being made to quantitate effects of anticancer agents on quality of life. Cancer drug clinical trials conventionally use a toxicity grading scale where grade 1 toxicities do not require treatment, grade 2 toxicities may require symptomatic treatment but are not life-threatening, grade 3 toxicities are potentially life-threatening if untreated, grade 4 toxicities are actually life-threatening, and grade 5 toxicities are those that result in the patient’s death.
Development of targeted agents may proceed quite differently. While phase 1–3 trials are still conducted, molecular analysis of human tumors may allow the precise definition of target expression in a patient’s tumor that is necessary for or relevant to the drug’s action. This information might then allow selection of patients expressing the drug target for participation in all trial phases. These patients may then have a greater chance of developing a useful response to the drug by virtue of expressing the target in the tumor. Clinical trials may be designed to incorporate an assessment of the behavior of the target in relation to the drug (pharmacodynamic studies). Ideally, the plasma concentration that affects the drug target is known, so escalation to MTD may not be necessary. Rather, the correlation of host toxicity while achieving an “optimal biologic dose” becomes a more relevant endpoint for phase 1 and early phase 2 trials with targeted agents.
Useful cancer drug treatment strategies using conventional chemotherapy agents, targeted agents, hormonal treatments, or biologics have one of two valuable outcomes. They can induce cancer cell death, resulting in tumor shrinkage with corresponding improvement in patient survival, or increase the time until the disease progresses. Another potential outcome is to induce cancer cell differentiation or dormancy with loss of tumor cell replicative potential and reacquisition of phenotypic properties resembling normal cells. A blocking in normal cellular differentiation may be a key feature in the pathogenesis of certain leukemias.
Cell death is a closely regulated process. Necrosis refers to cell death induced, for example, by physical damage with the hallmarks of cell swelling and membrane disruption. Apoptosis, or programmed cell death, refers to a highly ordered process whereby cells respond to defined stimuli by dying, and it recapitulates the necessary cell death observed during the ontogeny of the organism. Cancer chemotherapeutic agents can cause both necrosis and apoptosis. Apoptosis is characterized by chromatin condensation (giving rise to “apoptotic bodies”), cell shrinkage, and, in living animals, phagocytosis by surrounding stromal cells without evidence of inflammation. This process is regulated either by signal transduction systems that promote a cell’s demise after a certain level of insult is achieved or in response to specific cell-surface receptors that mediate physiologic cell death responses, such as occurs in the developing organism or in the normal function of immune cells. Influencing apoptosis by manipulation of signal transduction pathways has emerged as a basis for understanding the actions of drugs and designing new strategies to improve their use. Autophagy is a cellular response to injury where the cell does not initially die but catabolizes itself in a way that can lead to loss of replicative potential. A general view of how cancer treatments work is that the interaction of a chemotherapeutic drug with its target induces a “cascade” of further signaling steps. These signals ultimately lead to cell death by triggering an “execution phase” where proteases, nucleases, and endogenous regulators of the cell death pathway are activated (Fig. 103e-3).
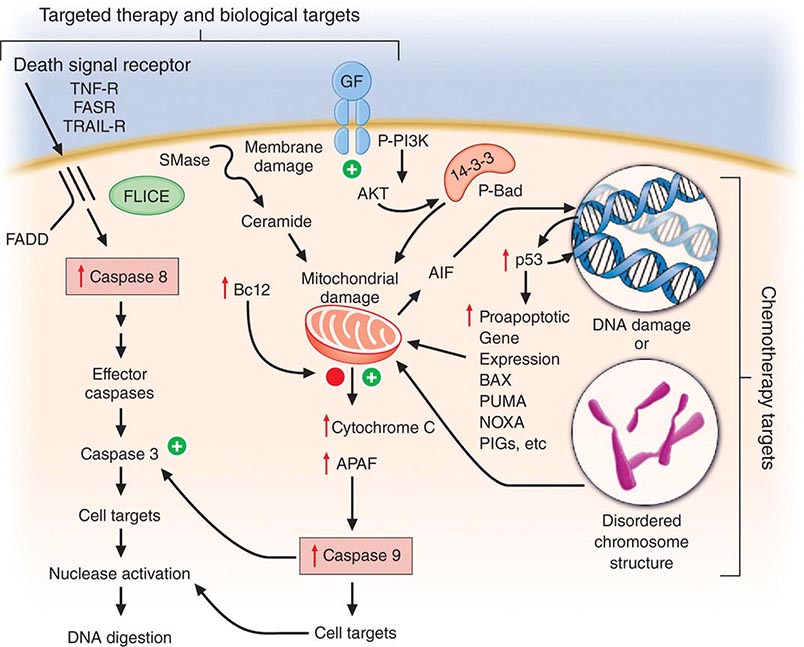
FIGURE 103e-3 Integration of cell death responses. Cell death through an apoptotic mechanism requires active participation of the cell. In response to interruption of growth factor (GF) or propagation of certain cytokine death signals (e.g., tumor necrosis factor receptor [TNF-R]), there is activation of “upstream” cysteine aspartyl proteases (caspases), which then directly digest cytoplasmic and nuclear proteins, resulting in activation of “downstream” caspases; these cause activation of nucleases, resulting in the characteristic DNA fragmentation that is a hallmark of apoptosis. Chemotherapy agents that create lesions in DNA or alter mitotic spindle function seem to activate aspects of this process by damage ultimately conveyed to the mitochondria, perhaps by activating the transcription of genes whose products can produce or modulate the toxicity of free radicals. In addition, membrane damage with activation of sphingomyelinases results in the production of ceramides that can have a direct action at mitochondria. The antiapoptotic protein bcl2 attenuates mitochondrial toxicity, while proapoptotic gene products such as bax antagonize the action of bcl2. Damaged mitochondria release cytochrome C and apoptosis-activating factor (APAF), which can directly activate caspase 9, resulting in propagation of a direct signal to other downstream caspases through protease activation. Apoptosis-inducing factor (AIF) is also released from the mitochondrion and then can translocate to the nucleus, bind to DNA, and generate free radicals to further damage DNA. An additional proapoptotic stimulus is the bad protein, which can heterodimerize with bcl2 gene family members to antagonize apoptosis. Importantly, though, bad protein function can be retarded by its sequestration as phospho-bad through the 14-3-3 adapter proteins. The phosphorylation of bad is mediated by the action of the AKT kinase in a way that defines how growth factors that activate this kinase can retard apoptosis and promote cell survival.
Targeted agents differ from chemotherapy agents in that they do not indiscriminately cause macromolecular lesions but regulate the action of particular pathways. For example, the p210bcr-abl fusion protein tyrosine kinase drives chronic myeloid leukemia (CML), and HER2/neu stimulates the proliferation of certain breast cancers. The tumor has been described as “addicted” to the function of these molecules in the sense that without the pathway’s continued action, the tumor cell cannot survive. In this way, targeted agents directed at p210bcr-abl or HER2/neu may alter the “threshold” tumors driven by these molecules may have for undergoing apoptosis without actually creating any molecular lesions such as direct DNA strand breakage or altered membrane function.
While apoptotic mechanisms are important in regulating cellular proliferation and the behavior of tumor cells in vitro, in vivo it is unclear whether all of the actions of chemotherapeutic agents to cause cell death can be attributed to apoptotic mechanisms. However, changes in molecules that regulate apoptosis are correlated with clinical outcomes (e.g., bcl2 overexpression in certain lymphomas conveys poor prognosis; proapoptotic bax expression is associated with a better outcome after chemotherapy for ovarian carcinoma). A better understanding of the relationship of cell death and cell survival mechanisms is needed.
Chemotherapy agents may be used for the treatment of active, clinically apparent cancer. The goal of such treatment in some cases is cure of the cancer, that is, elimination of all clinical and pathologic evidence of cancer and return of the patient to an expected survival no different than the general population. Table 103e-3, A lists those tumors considered curable by conventionally available chemotherapeutic agents when used to address disseminated or metastatic cancers. If a tumor is localized to a single site, serious consideration of surgery or primary radiation therapy should be given, because these treatment modalities may be curative as local treatments. Chemotherapy may then be used after the failure of these modalities to eradicate a local tumor or as part of multimodality approaches to offer primary treatment to a clinically localized tumor. In this event, it can allow organ preservation when given with radiation, as in the larynx or other upper airway sites, or sensitize tumors to radiation when given, e.g., to patients concurrently receiving radiation for lung or cervix cancer (Table 103e-3, B). Chemotherapy can be administered as an adjuvant, i.e., in addition to surgery or radiation (Table 103e-3, C), even after all clinically apparent disease has been removed. This use of chemotherapy has curative potential in breast and colorectal neoplasms, as it attempts to eliminate clinically unapparent tumor that may have already disseminated. As noted above, small tumors frequently have high growth fractions and therefore may be intrinsically more susceptible to the action of antiproliferative agents. Neoadjuvant chemotherapy refers to administration of chemotherapy prior to any surgery or radiation to a local tumor in an effort to enhance the effect of the local treatment.
CURABILITY OF CANCERS WITH CHEMOTHERAPY |

Chemotherapy is routinely used in “conventional” dose regimens. In general, these doses produce reversible acute side effects, primarily consisting of transient myelosuppression with or without gastrointestinal toxicity (usually nausea), which are readily managed. “High-dose” chemotherapy regimens are predicated on the observation that the dose-response curve for many anticancer agents is rather steep, and increased dose can produce markedly increased therapeutic effect, although at the cost of potentially life-threatening complications that require intensive support, usually in the form of hematopoietic stem cell support from the patient (autologous) or from donors matched for histocompatibility loci (allogeneic), or pharmacologic “rescue” strategies to repair the effect of the high-dose chemotherapy on normal tissues. High-dose regimens have definite curative potential in defined clinical settings (Table 103e-3, D).
If cure is not possible, chemotherapy may be undertaken with the goal of palliating some aspect of the tumor’s effect on the host. In this usage, value is perceived by the demonstration of improved symptom relief, progression-free survival, or overall survival at a certain time from the inception of treatment in the treated population, compared to a relevant control population established as the result of clinical research protocol or other organized comparative study. Such clinical research protocols are the basis for U.S. Food and Drug Administration (FDA) approval of a particular cancer treatment as safe and effective and are the benchmark for an evidence-based approach to the use of chemotherapeutic agents. Common tumors that may be meaningfully addressed by chemotherapy with palliative intent are listed in Table 103e-3, E.
Usually, tumor-related symptoms manifest as pain, weight loss, or some local symptom related to the tumor’s effect on normal structures. Patients treated with palliative intent should be aware of their diagnosis and the limitations of the proposed treatments, have access to supportive care, and have suitable “performance status,” according to assessment algorithms such as the one developed by Karnofsky (see Table 99-4) or by the Eastern Cooperative Oncology Group (ECOG) (see Table 99-5). ECOG performance status 0 (PS0) patients are without symptoms; PS1 patients are ambulatory but restricted in strenuous physical activity; PS2 patients are ambulatory but unable to work and are up and about 50% or more of the time; PS3 patients are capable of limited self-care and are up <50% of the time; and PS4 patients are totally confined to bed or chair and incapable of self-care. Only PS0, PS1, and PS2 patients are generally considered suitable for palliative (noncurative) treatment. If there is curative potential, even poor–performance status patients may be treated, but their prognosis is usually inferior to that of good–performance status patients treated with similar regimens.
An important perspective the primary care provider may bring to patients and their families facing incurable cancer is that, given the limited value of chemotherapeutic approaches at some point in the natural history of most metastatic cancers, palliative care or hospice-based approaches, with meticulous and ongoing attention to symptom relief and with family, psychological, and spiritual support, should receive prominent attention as a valuable therapeutic plan (Chaps. 10 and 99). Optimizing the quality of life rather than attempting to extend it becomes a valued intervention. Patients facing the impending progression of disease in a life-threatening way frequently choose to undertake toxic treatments of little to no potential value, and support provided by the primary caregiver in accessing palliative and hospice-based options in contrast to receiving toxic and ineffective regimen can be critical in providing a basis for patients to make sensible choices.
Cytotoxic Chemotherapy Agents Table 103e-4 lists commonly used cytotoxic cancer chemotherapy agents and pertinent clinical aspects of their use, with particular reference to adverse effects that might be encountered by the generalist in the care of patients. The drugs listed may be usefully grouped into two general categories: those affecting DNA and those affecting microtubules.
CYTOTOXIC CHEMOTHERAPY AGENTS |
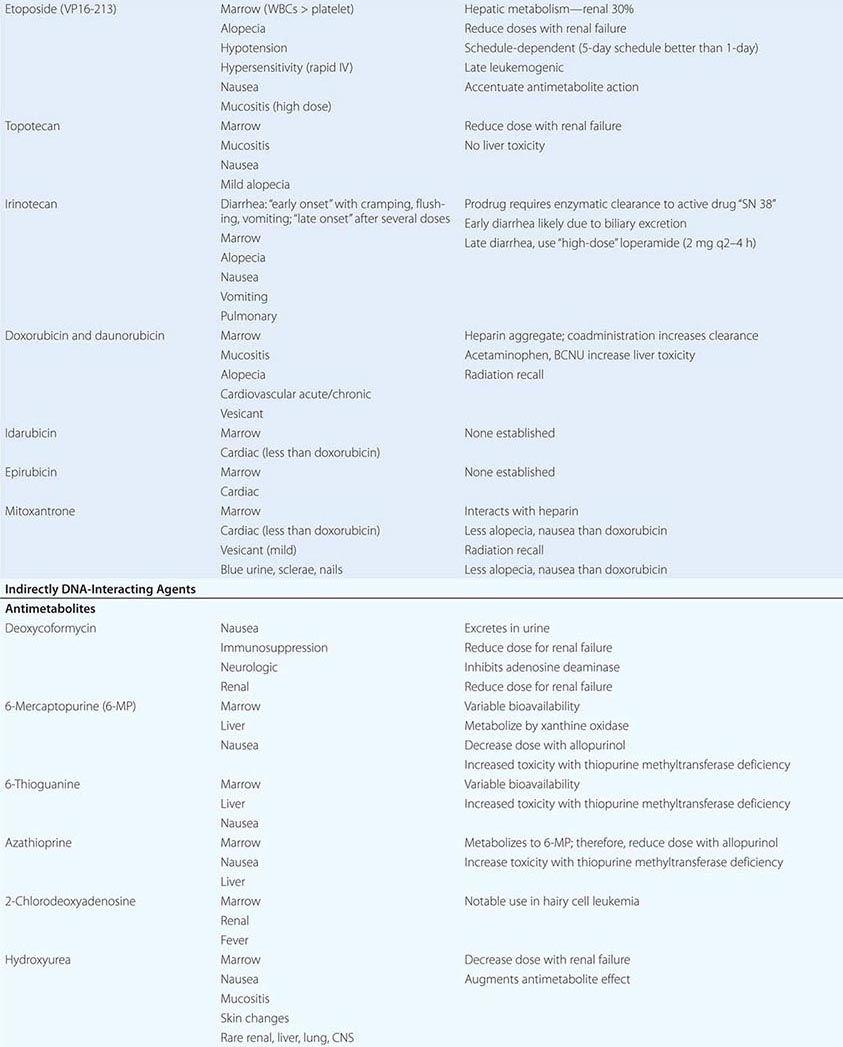
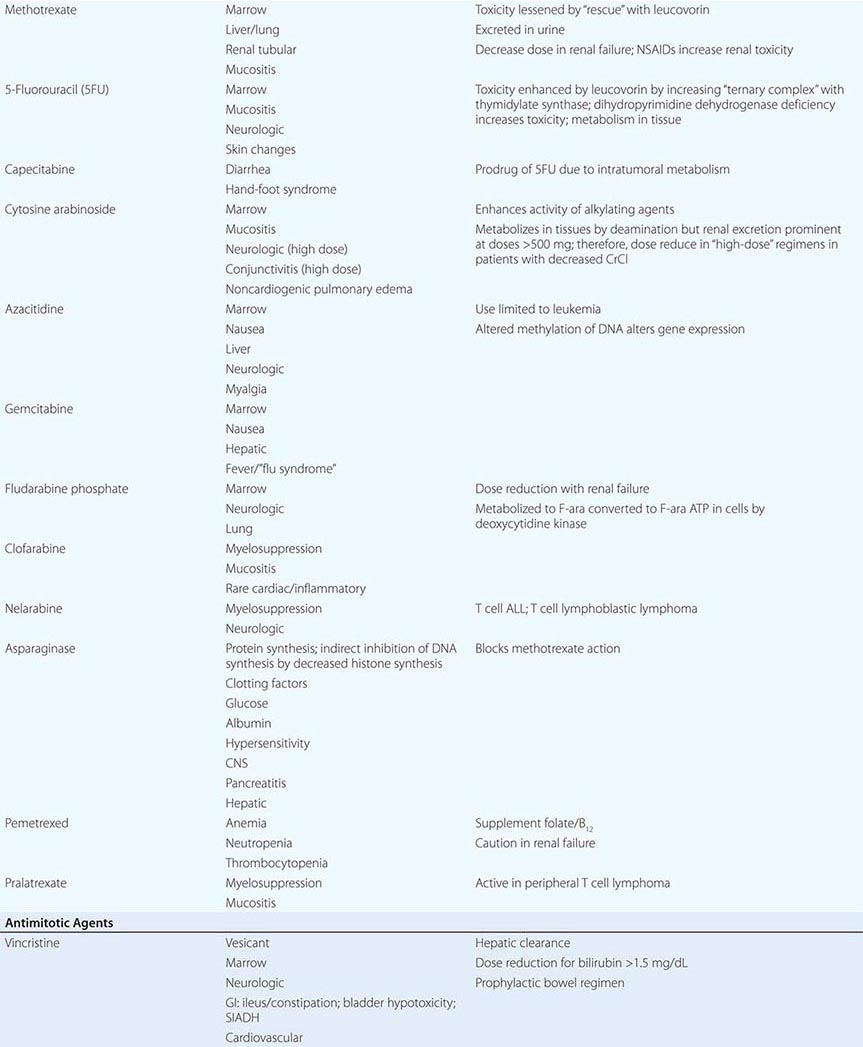
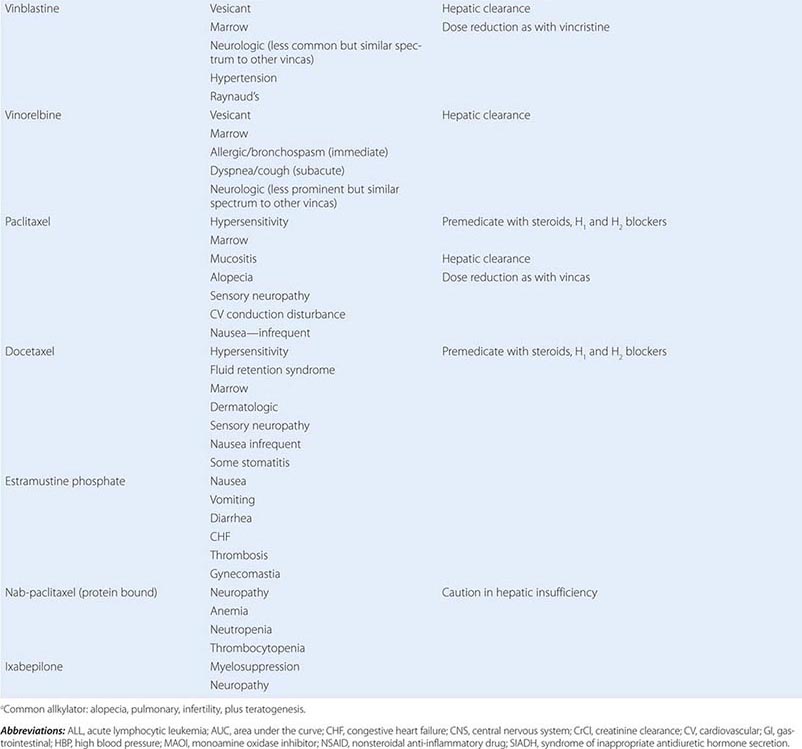
DIRECT DNA-INTERACTIVE AGENTS DNA replication occurs during the synthesis or S-phase of the cell cycle, with chromosome segregation of the replicated DNA occurring in the M, or mitosis, phase. The G1 and G2 “gap phases” precede S and M, respectively. Historically, chemotherapeutic agents have been divided into “phase-nonspecific” agents, which can act in any phase of the cell cycle, and “phase-specific” agents, which require the cell to be at a particular cell cycle phase to cause greatest effect. Once the agent has acted, cells may progress to “checkpoints” in the cell cycle where the drug-related damage may be assessed and either repaired or allowed to initiate apoptosis. An important function of certain tumor-suppressor genes such as p53 may be to modulate checkpoint function.
Alkylating agents as a class are cell cycle phase–nonspecific agents. They break down, either spontaneously or after normal organ or tumor cell metabolism, to reactive intermediates that covalently modify bases in DNA. This leads to cross-linkage of DNA strands or the appearance of breaks in DNA as a result of repair efforts. “Broken” or cross-linked DNA is intrinsically unable to complete normal replication or cell division; in addition, it is a potent activator of cell cycle checkpoints and further activates cell-signaling pathways that can precipitate apoptosis. As a class, alkylating agents share similar toxicities: myelosuppression, alopecia, gonadal dysfunction, mucositis, and pulmonary fibrosis. They differ greatly in a spectrum of normal organ toxicities. As a class, they share the capacity to cause “second” neoplasms, particularly leukemia, many years after use, particularly when used in low doses for protracted periods.
Cyclophosphamide is inactive unless metabolized by the liver to 4-hydroxy-cyclophosphamide, which decomposes into an alkylating species, as well as to chloroacetaldehyde and acrolein. The latter causes chemical cystitis; therefore, excellent hydration must be maintained while using cyclophosphamide. If severe, the cystitis may be prevented from progressing or prevented altogether (if expected from the dose of cyclophosphamide to be used) by mesna (2-mercaptoethanesulfonate). Liver disease impairs cyclophosphamide activation. Sporadic interstitial pneumonitis leading to pulmonary fibrosis can accompany the use of cyclophosphamide, and high doses used in conditioning regimens for bone marrow transplant can cause cardiac dysfunction. Ifosfamide is a cyclophosphamide analogue also activated in the liver, but more slowly, and it requires coadministration of mesna to prevent bladder injury. Central nervous system (CNS) effects, including somnolence, confusion, and psychosis, can follow ifosfamide use; the incidence appears related to low body surface area or decreased creatinine clearance.
Several alkylating agents are less commonly used. Nitrogen mustard (mechlorethamine) is the prototypic agent of this class, decomposing rapidly in aqueous solution to potentially yield a bifunctional carbonium ion. It must be administered shortly after preparation into a rapidly flowing intravenous line. It is a powerful vesicant, and infiltration may be symptomatically ameliorated by infiltration of the affected site with 1/6 M thiosulfate. Even without infiltration, aseptic thrombophlebitis is frequent. It can be used topically as a dilute solution or ointment in cutaneous lymphomas, with a notable incidence of hypersensitivity reactions. It causes moderate nausea after intravenous administration. Bendamustine is a nitrogen mustard derivative with evidence of activity in chronic lymphocytic leukemia and certain lymphomas.
Chlorambucil causes predictable myelosuppression, azoospermia, nausea, and pulmonary side effects. Busulfan can cause profound myelosuppression, alopecia, and pulmonary toxicity but is relatively “lymphocyte sparing.” Its routine use in treatment of CML has been curtailed in favor of imatinib (Gleevec) or dasatinib, but it is still used in transplant preparation regimens. Melphalan shows variable oral bioavailability and undergoes extensive binding to albumin and α1-acidic glycoprotein. Mucositis appears more prominently; however, it has prominent activity in multiple myeloma.
Nitrosoureas break down to carbamylating species that not only cause a distinct pattern of DNA base pair–directed toxicity but also can covalently modify proteins. They share the feature of causing relatively delayed bone marrow toxicity, which can be cumulative and long-lasting. Procarbazine is metabolized in the liver and possibly in tumor cells to yield a variety of free radical and alkylating species. In addition to myelosuppression, it causes hypnotic and other CNS effects, including vivid nightmares. It can cause a disulfiram-like syndrome on ingestion of ethanol. Altretamine (formerly hexa-methylmelamine) and thiotepa can chemically give rise to alkylating species, although the nature of the DNA damage has not been well characterized in either case. Dacarbazine (DTIC) is activated in the liver to yield the highly reactive methyl diazonium cation. It causes only modest myelosuppression 21–25 days after a dose but causes prominent nausea on day 1. Temozolomide is structurally related to dacarbazine but was designed to be activated by nonenzymatic hydrolysis in tumors and is bioavailable orally.
Cisplatin was discovered fortuitously by observing that bacteria present in electrolysis solutions with platinum electrodes could not divide. Only the cis diamine configuration is active as an antitumor agent. It is hypothesized that in the intracellular environment, a chloride is lost from each position, being replaced by a water molecule. The resulting positively charged species is an efficient bifunctional interactor with DNA, forming Pt-based cross-links. Cisplatin requires administration with adequate hydration, including forced diuresis with mannitol to prevent kidney damage; even with the use of hydration, gradual decrease in kidney function is common, along with noteworthy anemia. Hypomagnesemia frequently attends cisplatin use and can lead to hypocalcemia and tetany. Other common toxicities include neurotoxocity with stocking-and-glove sensorimotor neuropathy. Hearing loss occurs in 50% of patients treated with conventional doses. Cisplatin is intensely emetogenic, requiring prophylactic antiemetics. Myelosuppression is less evident than with other alkylating agents. Chronic vascular toxicity (Raynaud’s phenomenon, coronary artery disease) is a more unusual toxicity. Carboplatin displays less nephro-, oto-, and neurotoxicity. However, myelosuppression is more frequent, and because the drug is exclusively cleared through the kidney, adjustment of dose for creatinine clearance must be accomplished through use of various dosing nomograms. Oxaliplatin is a platinum analogue with noteworthy activity in colon cancers refractory to other treatments. It is prominently neurotoxic.
ANTITUMOR ANTIBIOTICS AND TOPOISOMERASE POISONS Antitumor antibiotics are substances produced by bacteria that in nature appear to provide a chemical defense against other hostile microorganisms. As a class, they bind to DNA directly and can frequently undergo electron transfer reactions to generate free radicals in close proximity to DNA, leading to DNA damage in the form of single-strand breaks or cross-links. Topoisomerase poisons include natural products or semisynthetic species derived ultimately from plants, and they modify enzymes that regulate the capacity of DNA to unwind to allow normal replication or transcription. These include topoisomerase I, which creates single-strand breaks that then rejoin following the passage of the other DNA strand through the break. Topoisomerase II creates double-strand breaks through which another segment of DNA duplex passes before rejoining. DNA damage from these agents can occur in any cell cycle phase, but cells tend to arrest in S-phase or G2 of the cell cycle in cells with p53 and Rb pathway lesions as the result of defective checkpoint mechanisms in cancer cells. Owing to the role of topoisomerase I in the procession of the replication fork, topoisomerase I poisons cause lethality if the topoisomerase I–induced lesions are made in S-phase.
Doxorubicin can intercalate into DNA, thereby altering DNA structure, replication, and topoisomerase II function. It can also undergo reduction reactions by accepting electrons into its quinone ring system, with the capacity to undergo reoxidation to form reactive oxygen radicals after reoxidation. It causes predictable myelosuppression, alopecia, nausea, and mucositis. In addition, it causes acute cardiotoxicity in the form of atrial and ventricular dysrhythmias, but these are rarely of clinical significance. In contrast, cumulative doses >550 mg/m2 are associated with a 10% incidence of chronic cardiomyopathy. The incidence of cardiomyopathy appears to be related to schedule (peak serum concentration), with low-dose, frequent treatment or continuous infusions better tolerated than intermittent higher-dose exposures. Cardiotoxicity has been related to iron-catalyzed oxidation and reduction of doxorubicin, and not to topoisomerase action. Cardiotoxicity is related to peak plasma dose; thus, lower doses and continuous infusions are less likely to cause heart damage. Doxorubicin’s cardiotoxicity is increased when given together with trastuzumab (Herceptin), the anti-HER2/neu antibody. Radiation recall or interaction with concomitantly administered radiation to cause local site complications is frequent. The drug is a powerful vesicant, with necrosis of tissue apparent 4–7 days after an extravasation; therefore, it should be administered into a rapidly flowing intravenous line. Dexrazoxane is an antidote to doxorubicin-induced extravasation. Doxorubicin is metabolized by the liver, so doses must be reduced by 50–75% in the presence of liver dysfunction. Daunorubicin is closely related to doxorubicin and was actually introduced first into leukemia treatment, where it remains part of curative regimens and has been shown preferable to doxorubicin owing to less mucositis and colonic damage. Idarubicin is also used in acute myeloid leukemia treatment and may be preferable to daunorubicin in activity. Encapsulation of daunorubicin into a liposomal formulation has attenuated cardiac toxicity and antitumor activity in Kaposi’s sarcoma, other sarcomas, multiple myeloma, and ovarian cancer.
Bleomycin refers to a mixture of glycopeptides that have the unique feature of forming complexes with Fe2+ while also bound to DNA. It remains an important component of curative regimens for Hodgkin’s disease and germ cell neoplasms. Oxidation of Fe2+ gives rise to superoxide and hydroxyl radicals. The drug causes little, if any, myelosuppression. The drug is cleared rapidly, but augmented skin and pulmonary toxicity in the presence of renal failure has led to the recommendation that doses be reduced by 50–75% in the face of a creatinine clearance <25 mL/min. Bleomycin is not a vesicant and can be administered intravenously, intramuscularly, or subcutaneously. Common side effects include fever and chills, facial flush, and Raynaud’s phenomenon. Hypertension can follow rapid intravenous administration, and the incidence of anaphylaxis with early preparations of the drug has led to the practice of administering a test dose of 0.5–1 unit before the rest of the dose. The most feared complication of bleomycin treatment is pulmonary fibrosis, which increases in incidence at >300 cumulative units administered and is minimally responsive to treatment (e.g., glucocorticoids). The earliest indicator of an adverse effect is usually a decline in the carbon monoxide diffusing capacity (DLco) or coughing, although cessation of drug immediately upon documentation of a decrease in DLco may not prevent further decline in pulmonary function. Bleomycin is inactivated by a bleomycin hydrolase, whose concentration is diminished in skin and lung. Because bleomycin-dependent electron transport is dependent on O2, bleomycin toxicity may become apparent after exposure to transient very high fraction of inspired oxygen (FIO2). Thus, during surgical procedures, patients with prior exposure to bleomycin should be maintained on the lowest FIo2 consistent with maintaining adequate tissue oxygenation.
Mitoxantrone is a synthetic compound that was designed to recapitulate features of doxorubicin but with less cardiotoxicity. It is quantitatively less cardiotoxic (comparing the ratio of cardiotoxic to therapeutically effective doses) but is still associated with a 10% incidence of cardiotoxicity at cumulative doses of >150 mg/m2. It also causes alopecia. Cases of acute promyelocytic leukemia (APL) have arisen shortly after exposure of patients to mitoxantrone, particularly in the adjuvant treatment of breast cancer. Although chemotherapy-associated leukemia is generally of the acute myeloid type, APL arising in the setting of prior mitoxantrone treatment had the typical t(15;17) chromosome translocation associated with APL, but the breakpoints of the translocation appeared to be at topoisomerase II sites that would be preferred sites of mitoxantrone action, clearly linking the action of the drug to the generation of the leukemia.
Etoposide was synthetically derived from the plant product podophyllotoxin; it binds directly to topoisomerase II and DNA in a reversible ternary complex. It stabilizes the covalent intermediate in the enzyme’s action where the enzyme is covalently linked to DNA. This “alkali-labile” DNA bond was historically a first hint that an enzyme such as a topoisomerase might exist. The drug therefore causes a prominent G2 arrest, reflecting the action of a DNA damage checkpoint. Prominent clinical effects include myelosuppression, nausea, and transient hypotension related to the speed of administration of the agent. Etoposide is a mild vesicant but is relatively free from other large-organ toxicities. When given at high doses or very frequently, topoisomerase II inhibitors may cause acute leukemia associated with chromosome 11q23 abnormalities in up to 1% of exposed patients.
Camptothecin was isolated from extracts of a Chinese tree and had notable antileukemia activity in preclinical mouse models. Early human clinical studies with the sodium salt of the hydrolyzed camptothecin lactone showed evidence of toxicity with little antitumor activity. Identification of topoisomerase I as the target of camptothecins and the need to preserve lactone structure allowed additional efforts to identify active members of this series. Topoisomerase I is responsible for unwinding the DNA strand by introducing single-strand breaks and allowing rotation of one strand about the other. In S-phase, topoisomerase I–induced breaks that are not promptly resealed lead to progress of the replication fork off the end of a DNA strand. The DNA damage is a potent signal for induction of apoptosis. Camptothecins promote the stabilization of the DNA linked to the enzyme in a so-called cleavable complex, analogous to the action of etoposide with topoisomerase II. Topotecan is a camptothecin derivative approved for use in gynecologic tumors and small-cell lung cancer. Toxicity is limited to myelosuppression and mucositis. CPT-11, or irinotecan, is a camptothecin with evidence of activity in colon carcinoma. In addition to myelosuppression, it causes a secretory diarrhea related to the toxicity of a metabolite called SN-38. Levels of SN-38 are particularly high in the setting of Gilbert’s disease, characterized by defective glucuronyl transferase and indirect hyperbilirubinemia, a condition that affects about 10% of the white population in the United States. The diarrhea can be treated effectively with loperamide or octreotide.
INDIRECT MODULATORS OF NUCLEIC ACID FUNCTION: ANTIMETABOLITES A broad definition of antimetabolites would include compounds with structural similarity to precursors of purines or pyrimidines, or compounds that interfere with purine or pyrimidine synthesis. Some antimetabolites can cause DNA damage indirectly, through misincorporation into DNA, abnormal timing or progression through DNA synthesis, or altered function of pyrimidine and purine biosynthetic enzymes. They tend to convey greatest toxicity to cells in S-phase, and the degree of toxicity increases with duration of exposure. Common toxic manifestations include stomatitis, diarrhea, and myelosuppression. Second malignancies are not associated with their use.
Methotrexate inhibits dihydrofolate reductase, which regenerates reduced folates from the oxidized folates produced when thymidine monophosphate is formed from deoxyuridine monophosphate. Without reduced folates, cells die a “thymine-less” death. N5-Tetrahydrofolate or N5-formyltetrahydrofolate (leucovorin) can bypass this block and rescue cells from methotrexate, which is maintained in cells by polyglutamylation. The drug and other reduced folates are transported into cells by the folate carrier, and high concentrations of drug can bypass this carrier and allow diffusion of drug directly into cells. These properties have suggested the design of “high-dose” methotrexate regimens with leucovorin rescue of normal marrow and mucosa as part of curative approaches to osteosarcoma in the adjuvant setting and hematopoietic neoplasms of children and adults. Methotrexate is cleared by the kidney via both glomerular filtration and tubular secretion, and toxicity is augmented by renal dysfunction and drugs such as salicylates, probenecid, and nonsteroidal anti-inflammatory agents that undergo tubular secretion. With normal renal function, 15 mg/m2 leucovorin will rescue 10–8 to 10–6 M methotrexate in three to four doses. However, with decreased creatinine clearance, doses of 50–100 mg/m2 are continued until methotrexate levels are <5 × 10–8 M. In addition to bone marrow suppression and mucosal irritation, methotrexate can cause renal failure itself at high doses owing to crystallization in renal tubules; therefore, high-dose regimens require alkalinization of urine with increased flow by hydration. Methotrexate can be sequestered in third-space collections and diffuse back into the general circulation, causing prolonged myelosuppression. Less frequent adverse effects include reversible increases in transaminases and hypersensitivity-like pulmonary syndrome. Chronic low-dose methotrexate can cause hepatic fibrosis. When administered to the intrathecal space, methotrexate can cause chemical arachnoiditis and CNS dysfunction.
Pemetrexed is a novel folate-directed antimetabolite. It is “multitargeted” in that it inhibits the activity of several enzymes, including thymidylate synthetase, dihydrofolate reductase, and glycinamide ribonucleotide formyltransferase, thereby affecting the synthesis of both purine and pyrimidine nucleic acid precursors. To avoid significant toxicity to the normal tissues, patients receiving pemetrexed should also receive low-dose folate and vitamin B12 supplementation. Pemetrexed has notable activity against certain lung cancers and, in combination with cisplatin, also against mesotheliomas. Pralatrexate is an antifolate approved for use in T cell lymphoma that is very efficiently transported into cancer cells.
5-Fluorouracil (5FU) represents an early example of “rational” drug design in that it originated from the observation that tumor cells incorporate radiolabeled uracil more efficiently into DNA than normal cells, especially gut. 5FU is metabolized in cells to 5´FdUMP, which inhibits thymidylate synthetase (TS). In addition, misincorporation can lead to single-strand breaks, and RNA can aberrantly incorporate FUMP. 5FU is metabolized by dihydropyrimidine dehydrogenase, and deficiency of this enzyme can lead to excessive toxicity from 5FU. Oral bioavailability varies unreliably, but orally administered analogues of 5FU such as capecitabine have been developed that allow at least equivalent activity to many parenteral 5FU-based approaches. Intravenous administration of 5FU leads to bone marrow suppression after short infusions but to stomatitis after prolonged infusions. Leucovorin augments the activity of 5FU by promoting formation of the ternary covalent complex of 5FU, the reduced folate, and TS. Less frequent toxicities include CNS dysfunction, with prominent cerebellar signs, and endothelial toxicity manifested by thrombosis, including pulmonary embolus and myocardial infarction.
Cytosine arabinoside (ara-C) is incorporated into DNA after formation of ara-CTP, resulting in S-phase–related toxicity. Continuous infusion schedules allow maximal efficiency, with uptake maximal at 5–7 μM. Ara-C can be administered intrathecally. Adverse effects include nausea, diarrhea, stomatitis, chemical conjunctivitis, and cerebellar ataxia. Gemcitabine is a cytosine derivative that is similar to ara-C in that it is incorporated into DNA after anabolism to the triphosphate, rendering DNA susceptible to breakage and repair synthesis, which differs from that in ara-C in that gemcitabine-induced lesions are very inefficiently removed. In contrast to ara-C, gemcitabine appears to have useful activity in a variety of solid tumors, with limited nonmyelosuppressive toxicities.
6-Thioguanine and 6-mercaptopurine (6MP) are used in the treatment of acute lymphoid leukemia. Although administered orally, they display variable bioavailability. 6MP is metabolized by xanthine oxidase and therefore requires dose reduction when used with allopurinol. 6MP is also metabolized by thiopurine methyltransferase; genetic deficiency of thiopurine methyltransferase results in excessive toxicity.
Fludarabine phosphate is a prodrug of F-adenine arabinoside (F-ara-A), which in turn was designed to diminish the susceptibility of ara-A to adenosine deaminase. F-ara-A is incorporated into DNA and can cause delayed cytotoxicity even in cells with low growth fraction, including chronic lymphocytic leukemia and follicular B cell lymphoma. CNS and peripheral nerve dysfunction and T cell depletion leading to opportunistic infections can occur in addition to myelosuppression. 2-Chlorodeoxyadenosine is a similar compound with activity in hairy cell leukemia. 2-Deoxycoformycin inhibits adenosine deaminase, with resulting increase in dATP levels. This causes inhibition of ribonucleotide reductase as well as augmented susceptibility to apoptosis, particularly in T cells. Renal failure and CNS dysfunction are notable toxicities in addition to immunosuppression. Hydroxyurea inhibits ribonucleotide reductase, resulting in S-phase block. It is orally bioavailable and useful for the acute management of myeloproliferative states.
Asparaginase is a bacterial enzyme that causes breakdown of extracellular asparagine required for protein synthesis in certain leukemic cells. This effectively stops tumor cell DNA synthesis, as DNA synthesis requires concurrent protein synthesis. The outcome of asparaginase action is therefore very similar to the result of the small-molecule antimetabolites. Because asparaginase is a foreign protein, hypersensitivity reactions are common, as are effects on organs such as pancreas and liver that normally require continuing protein synthesis. This may result in decreased insulin secretion with hyperglycemia, with or without hyperamylasemia and clotting function abnormalities. Close monitoring of clotting functions should accompany use of asparaginase. Paradoxically, owing to depletion of rapidly turning over anticoagulant factors, thromboses particularly affecting the CNS may also be seen with asparaginase.
MITOTIC SPINDLE INHIBITORS Microtubules are cellular structures that form the mitotic spindle, and in interphase cells, they are responsible for the cellular “scaffolding” along which various motile and secretory processes occur. Microtubules are composed of repeating noncovalent multimers of a heterodimer of α and β isoform of the protein tubulin. Vincristine binds to the tubulin dimer with the result that microtubules are disaggregated. This results in the block of growing cells in M-phase; however, toxic effects in G1 and S-phase are also evident, reflecting effects on normal cellular activities of microtubules. Vincristine is metabolized by the liver, and dose adjustment in the presence of hepatic dysfunction is required. It is a powerful vesicant, and infiltration can be treated by local heat and infiltration of hyaluronidase. At clinically used intravenous doses, neurotoxicity in the form of glove-and-stocking neuropathy is frequent. Acute neuropathic effects include jaw pain, paralytic ileus, urinary retention, and the syndrome of inappropriate antidiuretic hormone secretion. Myelosuppression is not seen. Vinblastine is similar to vincristine, except that it tends to be more myelotoxic, with more frequent thrombocytopenia and also mucositis and stomatitis. Vinorelbine is a vinca alkaloid that appears to have differences in resistance patterns in comparison to vincristine and vinblastine; it may be administered orally.
The taxanes include paclitaxel and docetaxel. These agents differ from the vinca alkaloids in that the taxanes stabilize microtubules against depolymerization. The “stabilized” microtubules function abnormally and are not able to undergo the normal dynamic changes of microtubule structure and function necessary for cell cycle completion. Taxanes are among the most broadly active antineoplastic agents for use in solid tumors, with evidence of activity in ovarian cancer, breast cancer, Kaposi’s sarcoma, and lung tumors. They are administered intravenously, and paclitaxel requires use of a Cremophor-containing vehicle that can cause hypersensitivity reactions. Premedication with dexamethasone (8–16 mg orally or intravenously 12 and 6 h before treatment) and diphenhydramine (50 mg) and cimetidine (300 mg), both 30 min before treatment, decreases but does not eliminate the risk of hypersensitivity reactions to the paclitaxel vehicle. Docetaxel uses a polysorbate 80 formulation, which can cause fluid retention in addition to hypersensitivity reactions, and dexamethasone premedication with or without antihistamines is frequently used. A protein-bound formulation of paclitaxel (called nab-paclitaxel) has at least equivalent antineoplastic activity and decreased risk of hypersensitivity reactions. Paclitaxel may also cause hypersensitivity reactions, myelosuppression, neurotoxicity in the form of glove-and-stocking numbness, and paresthesia. Cardiac rhythm disturbances were observed in phase 1 and 2 trials, most commonly asymptomatic bradycardia but also, much more rarely, varying degrees of heart block. These have not emerged as clinically significant in the majority of patients. Docetaxel causes comparable degrees of myelosuppression and neuropathy. Hypersensitivity reactions, including bronchospasm, dyspnea, and hypotension, are less frequent but occur to some degree in up to 25% of patients. Fluid retention appears to result from a vascular leak syndrome that can aggravate preexisting effusions. Rash can complicate docetaxel administration, appearing prominently as a pruritic maculopapular rash affecting the forearms, but it has also been associated with fingernail ridging, breakdown, and skin discoloration. Stomatitis appears to be somewhat more frequent than with paclitaxel. Cabazitaxel is a taxane with somewhat better activity in prostate cancers than earlier generations of taxanes, perhaps due to superior delivery to sites of disease.
Resistance to taxanes has been related to the emergence of efficient efflux of taxanes from tumor cells through the p170 P-glycoprotein (mdr gene product) or the presence of variant or mutant forms of tubulin. Epothilones represent a class of novel microtubule-stabilizing agents that have been conscientiously optimized for activity in taxane-resistant tumors. Ixabepilone has clear evidence of activity in breast cancers resistant to taxanes and anthracyclines such as doxorubicin. It retains acceptable expected side effects, including myelosuppression, and can also cause peripheral sensory neuropathy. Eribulin is a microtubule-directed agent with activity in patients who have had progression of disease on taxanes and is more similar to vinca alkaloids in its action but has similar side effects as vinca alkaloids and taxanes.
Estramustine was originally synthesized as a mustard derivative that might be useful in neoplasms that possessed estrogen receptors. However, no evidence of interaction with DNA was observed. Surprisingly, the drug caused metaphase arrest, and subsequent study revealed that it binds to microtubule-associated proteins, resulting in abnormal microtubule function. Estramustine binds to estramustine-binding proteins (EMBPs), which are notably present in prostate tumor tissue, where the drug is used. Gastrointestinal and cardiovascular adverse effects related to the estrogen moiety occur in up to 10% of patients, including worsened heart failure and thromboembolic phenomena. Gynecomastia and nipple tenderness can also occur.
Targeted Chemotherapy • HORMONE RECEPTOR–DIRECTED THERAPY Steroid hormone receptor–related molecules have emerged as prominent targets for small molecules useful in cancer treatment. When bound to their cognate ligands, these receptors can alter gene transcription and, in certain tissues, induce apoptosis. The pharmacologic effect is a mirror or parody of the normal effects of the agents acting on nontransformed normal tissues, although the effects on tumors are mediated by indirect effects in some cases. While in some cases, such as breast cancer, demonstration of the target hormone receptor is necessary, in other cases such prostate cancer (androgen receptor) and lymphoid neoplasms (glucocorticoid receptor), the relevant receptor is always present in the tumor.
Glucocorticoids are generally given in “pulsed” high doses in leukemias and lymphomas, where they induce apoptosis in tumor cells. Cushing’s syndrome and inadvertent adrenal suppression on withdrawal from high-dose glucocorticoids can be significant complications, along with infections common in immunosuppressed patients, in particular Pneumocystis pneumonia, which classically appears a few days after completing a course of high-dose glucocorticoids.
Tamoxifen is a partial estrogen receptor antagonist; it has a 10-fold greater antitumor activity in breast cancer patients whose tumors express estrogen receptors than in those who have low or no levels of expression. It might be considered the prototypic “molecularly targeted” agent. Owing to its agonistic activities in vascular and uterine tissue, side effects include a somewhat increased risk of cardiovascular complications, such as thromboembolic phenomena, and a small increased incidence of endometrial carcinoma, which appears after chronic use (usually >5 years). Progestational agents—including medroxyprogesterone acetate, androgens including fluoxymesterone (Halotestin), and, paradoxically, estrogens—have approximately the same degree of activity in primary hormonal treatment of breast cancers that have elevated expression of estrogen receptor protein. Estrogen itself is not used often owing to prominent cardiovascular and uterotropic activity.
Aromatase refers to a family of enzymes that catalyze the formation of estrogen in various tissues, including the ovary and peripheral adipose tissue and some tumor cells. Aromatase inhibitors are of two types, the irreversible steroid analogues such as exemestane and the reversible inhibitors such as anastrozole or letrozole. Anastrozole is superior to tamoxifen in the adjuvant treatment of breast cancer in postmenopausal patients with estrogen receptor–positive tumors. Letrozole treatment affords benefit following tamoxifen treatment. Adverse effects of aromatase inhibitors may include an increased risk of osteoporosis.
Prostate cancer is classically treated by androgen deprivation. Diethylstilbestrol (DES) acting as an estrogen at the level of the hypothalamus to downregulate hypothalamic luteinizing hormone (LH) production results in decreased elaboration of testosterone by the testicle. For this reason, orchiectomy is equally as effective as moderate-dose DES, inducing responses in 80% of previously untreated patients with prostate cancer but without the prominent cardiovascular side effects of DES, including thrombosis and exacerbation of coronary artery disease. In the event that orchiectomy is not accepted by the patient, testicular androgen suppression can also be effected by luteinizing hormone–releasing hormone (LHRH) agonists such as leuprolide and goserelin. These agents cause tonic stimulation of the LHRH receptor, with the loss of its normal pulsatile activation resulting in decreased output of LH by the anterior pituitary. Therefore, as primary hormonal manipulation in prostate cancer, one can choose orchiectomy or leuprolide, but not both. The addition of androgen receptor blockers, including flutamide or bicalutamide, is of uncertain additional benefit in extending overall response duration; the combined use of orchiectomy or leuprolide plus flutamide is referred to as total androgen blockade. Enzalutamide also binds to the androgen receptor and antagonizes androgen action in a mechanistically distinct way. Somewhat analogous to inhibitors of aromatase, agents have been derived that inhibit testosterone and other androgen synthesis in the testis, adrenal gland, and prostate tissue. Abiraterone inhibits 17 α-hydroxylase/C17,20 lyase (CYP 17A1) and has been shown to be active in prostate cancer patients experiencing progression despite androgen blockade.
Tumors that respond to a primary hormonal manipulation may frequently respond to second and third hormonal manipulations. Thus, breast tumors that had previously responded to tamoxifen have, on relapse, notable response rates to withdrawal of tamoxifen itself or to subsequent addition of an aromatase inhibitor or progestin. Likewise, initial treatment of prostate cancers with leuprolide plus flutamide may be followed after disease progression by response to withdrawal of flutamide. These responses may result from the removal of antagonists from mutant steroid hormone receptors that have come to depend on the presence of the antagonist as a growth-promoting influence.
Additional strategies to treat refractory breast and prostate cancers that possess steroid hormone receptors may also address adrenal capacity to produce androgens and estrogens, even after orchiectomy or oophorectomy, respectively. Thus, aminoglutethimide or ketoconazole can be used to block adrenal synthesis by interfering with the enzymes of steroid hormone metabolism. Administration of these agents requires concomitant hydrocortisone replacement and additional glucocorticoid doses administered in the event of physiologic stress.
Humoral mechanisms can also result in complications from an underlying malignancy producing the hormone. Adrenocortical carcinomas can cause Cushing’s syndrome as well as syndromes of androgen or estrogen excess. Mitotane can counteract these by decreasing synthesis of steroid hormones. Islet cell neoplasms can cause debilitating diarrhea, treated with the somatostatin analogue octreotide. Prolactin-secreting tumors can be effectively managed by the dopaminergic agonist bromocriptine.
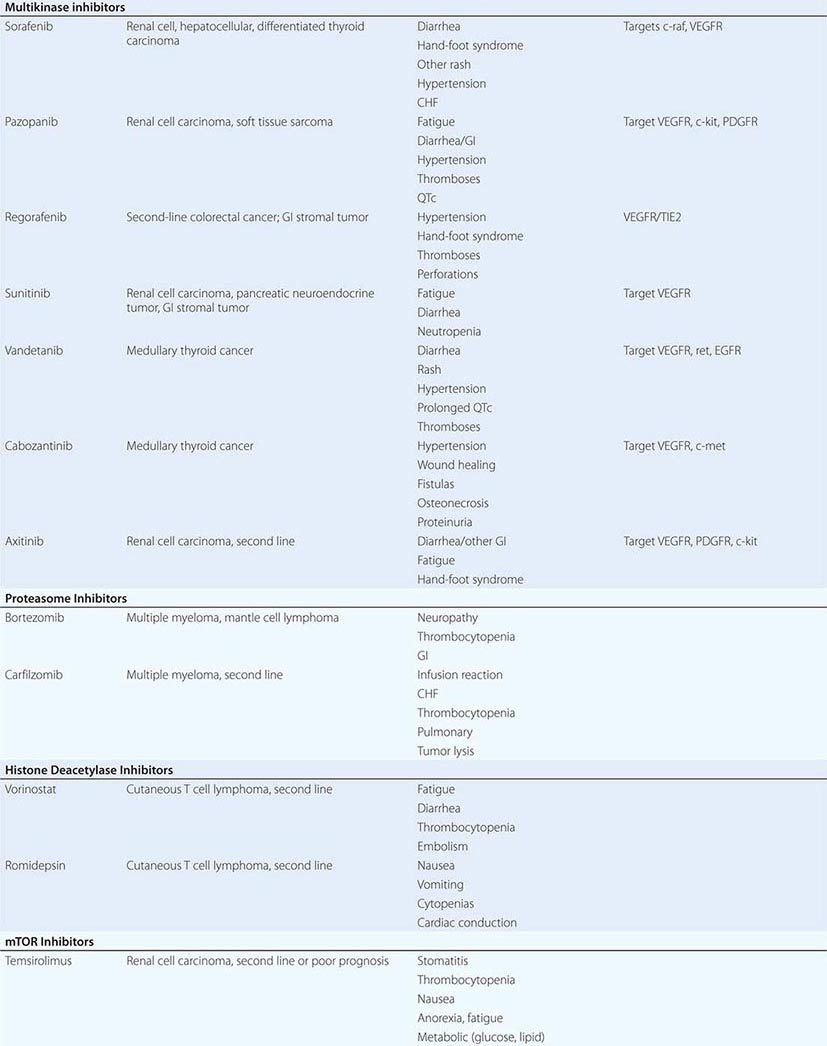
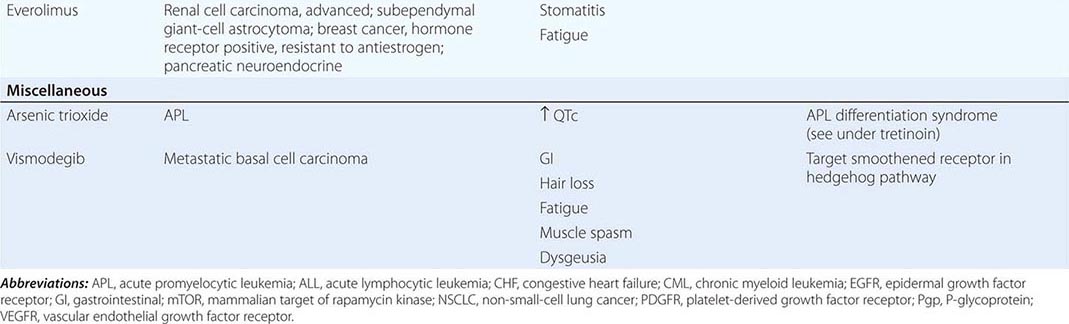
DIAGNOSTICALLY GUIDED THERAPY The basis for discovery of drugs of this type was the prior knowledge of the importance of the drugs’ molecular target to drive tumors in different contexts. Figure 103e-4 summarizes how FDA-approved targeted agents act. In the case of diagnostically guided targeted chemotherapy, prior demonstration of a specific target is necessary to guide the rational use of the agent, while in the case of targeted agents directed at oncogenic pathways, specific diagnosis of pathway activation is not yet necessary or in some cases feasible, although this is an area of ongoing clinical research. Table 103e-5 lists currently approved targeted chemotherapy agents, with features of their use.
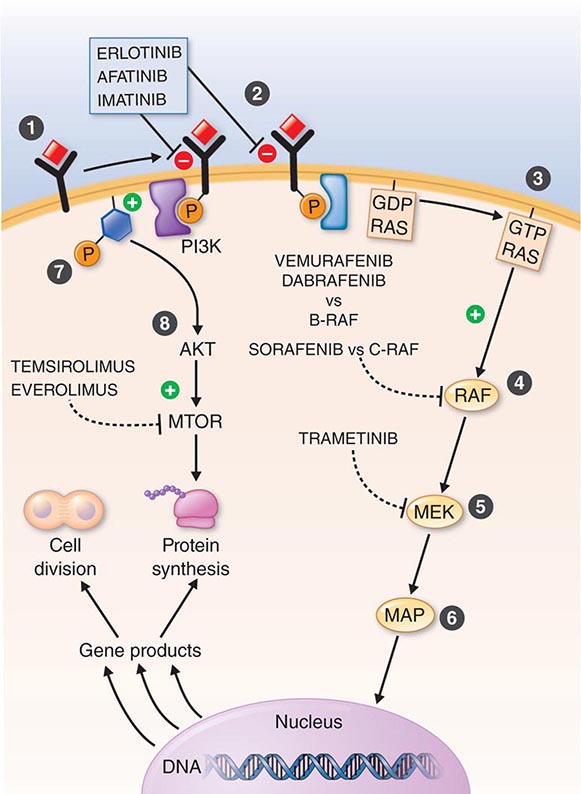
FIGURE 103e-4 Targeted chemotherapeutic agents act in most instances by interrupting cell growth factor-mediated signaling pathways. After a growth factor binds to is cognate receptor (1), in many cases there is activation of tyrsosine kinase activity particularly after dimerization of the receptors (2). This leads to autophosphorylation of the receptor and docking of “adaptor” proteins. One important pathway activated occurs after exchange of GDP for GTP in the RAS family of proto-oncogene products (3). GTP-RAS activates the RAF proto-oncogene kinase (4), leading to a phosphorylation cascade of kinases (5, 6) that ultimately impart signals to regulators of gene function to produce transcripts which activate cell cycle progression and increase protein synthesis. In parallel, tyrosine phosphorylated receptors can activate the phosphatidylinositol-3-kinase to produce the phosphorylated lipid phosphatidyl-inositol-3- phosphate (7). This leads to the activation of the AKT kinase(8) which in turn stimulates the mammalian “Target of Rapamycin” kinase (mTOR), which directly increases the translation of key mRNAs for gene products regulating cell growth. Erlotinib and afatinib, are examples of Epidermal Growth Factor receptor tyrosine kinase inhibitors; imatinib can act on the nonreceptor tyrosine kinase bcr-abl or c-KIT membrane bound tyrosine kinase. Vemurafenib and Dabrafenib act on the B isoform of RAF uniquely in melanoma, and c-RAF is inhibited by sorafenib. Trametinib acts on MEK. Temsirolimus and everolimus inhibit mTOR kinase to downregulate translation of oncogenic mRNAs.
MOLECULARLY TARGETED AGENTS |
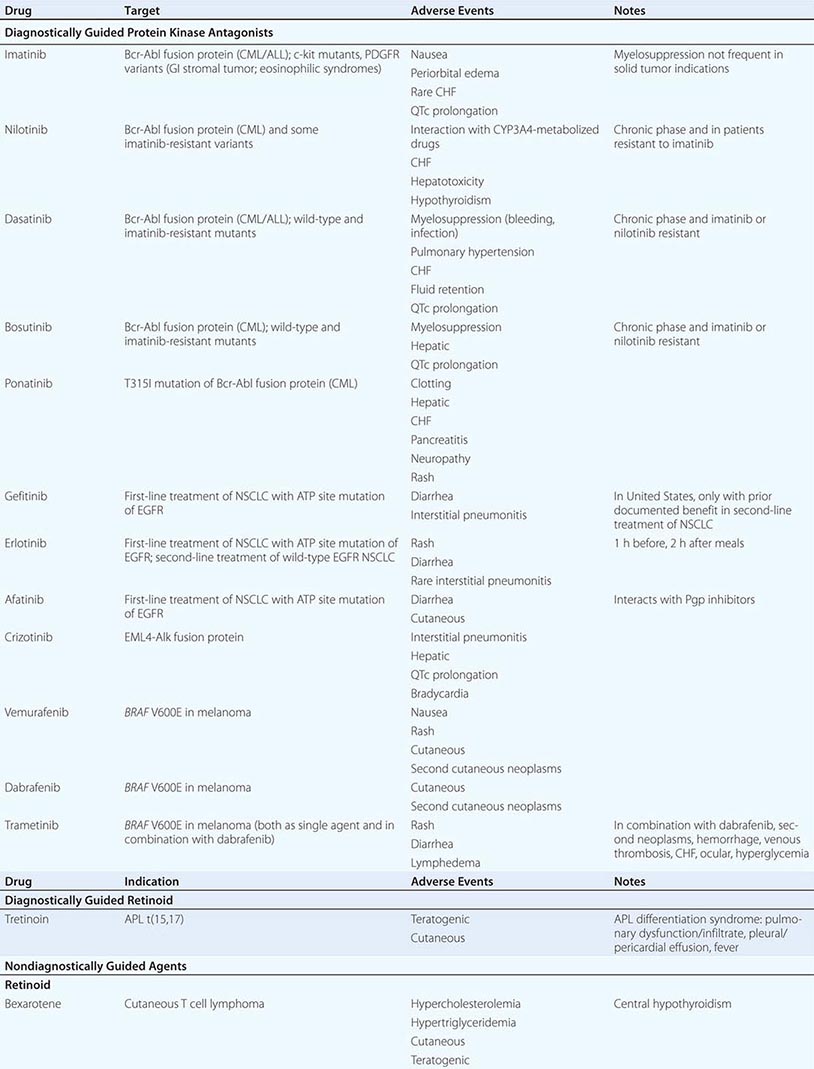
In hematologic tumors, the prototypic agent of this type is imatinib, which targets the ATP binding site of the p210bcr-abl protein tyrosine kinase that is formed as the result of the chromosome 9;22 translocation producing the Philadelphia chromosome in CML. Imatinib is superior to interferon plus chemotherapy in the initial treatment of the chronic phase of this disorder. It has lesser activity in the blast phase of CML, where the cells may have acquired additional mutations in p210bcr-abl itself or other genetic lesions. Its side effects are relatively tolerable in most patients and include hepatic dysfunction, diarrhea, and fluid retention. Rarely, patients receiving imatinib have decreased cardiac function, which may persist after discontinuation of the drug. The quality of response to imatinib enters into the decision about when to refer patients with CML for consideration of transplant approaches. Nilotinib is a tyrosine protein kinase inhibitor with a similar spectrum of activity to imatinib, but with increased potency and perhaps better tolerance by certain patients. Dasatinib, another inhibitor of the p210bcr-abl oncoproteins, is active in certain mutant variants of p210bcr-abl that are refractory to imatinib and arise during therapy with imatinib or are present de novo. Dasatinib also has inhibitory action against kinases belonging to the src tyrosine protein kinase family; this activity may contribute to its effects in hematopoietic tumors and suggest a role in solid tumors where src kinases are active. The T315I mutant of p210bcr-abl is resistant to imatinib, nilotinib, bosutinib, and dasatinib; ponatinib has activity in patients with this p210bcr-abl variant, but ponatinib has noteworthy associated thromboembolic toxicity. Use of this class of targeted agents is thus critically guided not only by the presence of the p210bcr-abl tyrosine kinase, but also by the presence of different mutations in the ATP binding site.
All-trans-retinoic acid (ATRA) targets the PML-retinoic acid receptor (RAR) α fusion protein, which is the result of the chromosome 15;17 translocation pathogenic for most forms of APL. Administered orally, it causes differentiation of the neoplastic promyelocytes to mature granulocytes and attenuates the rate of hemorrhagic complications. Adverse effects include headache with or without pseudotumor cerebri and gastrointestinal and cutaneous toxicities.
In epithelial solid tumors, the small-molecule epidermal growth factor (EGF) antagonists act at the ATP binding site of the EGF receptor tyrosine kinase. In early clinical trials, gefitinib showed evidence of responses in a small fraction of patients with non-small-cell lung cancer (NSCLC). Side effects were generally acceptable, consisting mostly of rash and diarrhea. Subsequent analysis of responding patients revealed a high frequency of activating mutations in the EGF receptor. Patients with such activating mutations who initially responded to gefitinib but who then had progression of the disease then acquired additional mutations in the enzyme, analogous functionally to mutational variants responsible for imatinib resistance in CML. Erlotinib is another EGF receptor tyrosine kinase antagonist with a superior outcome in clinical trials in NSCLC; an overall survival advantage was demonstrated in subsets of patients who were treated after demonstrating progression of disease and who also had not been preselected for the presence of activating mutations. Thus, although even patients with wild-type EGF receptors may benefit from erlotinib treatment, the presence of EGF receptor tyrosine kinase mutations has recently been shown to be a basis for recommending erlotinib and afatinib for first-line treatment of advanced NSCLC. Likewise, crizotinib targeting the alk protooncogene fusion protein has value in the initial treatment of alk-positive NSCLC. Lapatinib is a tyrosine kinase inhibitor with both EGF receptor and HER2/neu antagonist activity, which is important in the treatment of breast cancers expressing the HER2/neu oncoprotein.
In addition to the p210bcr-abl kinase, imatinib also has activity against the c-kit tyrosine kinase (the receptor for the steel growth factor, also called stem cell factor) and the platelet-derived growth factor receptor (PDGFR), both of which can be expressed in gastrointestinal stromal sarcoma (GIST). Imatinib has found clinical utility in GIST, a tumor previously notable for its refractoriness to chemotherapeutic approaches. Imatinib’s degree of activity varies with the specific mutational variant of kit or PDGFR present in a particular patient’s tumor.
The BRAF V600E mutation has been detected in a notable fraction of melanomas, thyroid tumors, and hairy cell leukemia, and preclinical models supported the concept that BRAF V600E drives oncogenic signaling in these tumors. Vemurafenib and dabrafenib, with selective capacity to inhibit the BRAF V600E serine kinase activity, were each shown to cause noteworthy responses in patients with BRAF V600E–mutated melanomas, although early relapse occurred in many patients treated with the drugs as single agents. Trametinib, acting downstream of BRAF V600E by directly inhibiting the MEK serine kinase by a non-ATP binding site mechanism, also displayed noteworthy responses in BRAF V600E–mutated melanomas, and the combination of trametinib and dabrafenib is even more active, by targeting the BRAF V600E–driven pathway at two points in the pathway leading to gene activation.
ONCOGENICALLY ACTIVATED PATHWAYS This group of agents also targets specific regulatory molecules in promoting the viability of tumor cells, but they do not require the diagnostically verified presence of a particular target or target variant at this time.
“Multitargeted” kinase antagonists are small-molecule ATP site-directed antagonists that inhibit more than one protein kinase and have value in the treatment of several solid tumors. Drugs of this type with prominent activity against the vascular endothelial growth factor receptor (VEGFR) tyrosine kinase have activity in renal cell carcinoma. Sorafenib is a VEGFR antagonist with activity against the raf serine-threonine protein kinase, and regorafenib is a closely related drug with value in relapsed advanced colon cancer. Pazopanib also prominently targets VEGFR and has activity in renal carcinoma and soft tissue sarcomas. Sunitinib has anti-VEGFR, anti-PDGFR, and anti-c-kit activity. It causes prominent responses and stabilization of disease in renal cell cancers and GISTs. Side effects for agents with anti-VEGFR activity prominently include hypertension, proteinuria, and, more rarely, bleeding and clotting disorders and perforation of scarred gastrointestinal lesions. Also encountered are fatigue, diarrhea, and the hand-foot syndrome, with erythema and desquamation of the distal extremities, in some cases requiring dose modification, particularly with sorafenib.
Temsirolimus and everolimus are mammalian target of rapamycin (mTOR) inhibitors with activity in renal cancers. They produce stomatitis, fatigue, and some hyperlipidemia (10%), myelosuppression (10%), and rare lung toxicity. Everolimus is also useful in patients with hormone receptor–positive breast cancers displaying resistance to hormonal inhibition and in certain neuroendocrine and brain tumors, the latter arising in patients with sporadic or inherited mutations in the pathway activating mTOR.
In hematologic neoplasms, bortezomib is an inhibitor of the proteasome, the multisubunit assembly of protease activities responsible for the selective degradation of proteins important in regulating activation of transcription factors, including nuclear factor-κB (NF-κB) and proteins regulating cell cycle progression. It has activity in multiple myeloma and certain lymphomas. Adverse effects include neuropathy, orthostatic hypotension with or without hyponatremia, and reversible thrombocytopenia. Carfilzomib is a proteasome inhibitor chemically unrelated to bortezomib without prominent neuropathy, but with evidence of a cytokine release syndrome, which can be a cardiopulmonary stress. Other agents active in multiple myeloma and certain other hematologic neoplasms include the immunomodulatory agents related to thalidomide, including lenalidomide and pomalidomide. All these agents collectively inhibit aberrant angiogenesis in the bone marrow microenvironment, as well as influence stromal cell immune functions to alter the cytokine milieu supporting the growth of myeloma cells. Thalidomide, although clinically active, has prominent cytopenic, neuropathic, procoagulant, and CNS toxicities that have been somewhat attenuated in the other drugs of the class, although use of these agents frequently entails concomitant anticoagulant prophylaxis.
Ibrutinib is representative a novel class of inhibitors directed at Bruton’s tyrosine kinase, which is important in the function of B cells. Initially approved for use in mantle cell lymphoma, it is potentially applicable to a number of B cell neoplasms that depend on signals through the B cell antigen receptor. Janus kinases likewise function downstream of a variety of cytokine receptors to amplify cytokine signals, and Janus kinase inhibitors including ruxolitinib have approved activity in myelofibrosis to ameliorate splenomegaly and systemic symptoms.
Vorinostat is an inhibitor of histone deacetylases, which are responsible for maintaining the proper orientation of histones on DNA, with resulting capacity for transcriptional readiness. Acetylated histones allow access of transcription factors to target genes and therefore increase expression of genes that are selectively repressed in tumors. The result can be differentiation with the emergence of a more normal cellular phenotype, or cell cycle arrest with expression of endogenous regulators of cell cycle progression. Vorinostat is approved for clinical use in cutaneous T cell lymphoma, with dramatic skin clearing and very few side effects. Romidepsin is a distinct molecular class of histone deacetylase inhibitor also active in cutaneous T cell lymphoma. An active retinoid in cutaneous T cell lymphoma is the synthetic retinoid × receptor ligand bexarotene.
DNA methyltransferase inhibitors, including 5-aza-cytidine and 2´-deoxy-5-azacytidine (decitabine), can also increase transcription of genes “silenced” during the pathogenesis of a tumor by causing demethylation of the methylated cytosines that are acquired as an “epigenetic” (i.e., after the DNA is replicated) modification of DNA. These drugs were originally considered antimetabolites but have clinical value in myelodysplastic syndromes and certain leukemias when administered at low doses.
CANCER BIOLOGIC THERAPY
Principles The goal of biologic therapy is to manipulate the host–tumor interaction in favor of the host, potentially at an optimum biologic dose that might be different than the MTD. As a class, biologic therapies may be distinguished from molecularly targeted agents in that many biologic therapies require an active response (e.g., reexpression of silenced genes or antigen expression) on the part of the tumor cell or on the part of the host (e.g., immunologic effects) to allow therapeutic effect. This may be contrasted with the more narrowly defined antiproliferative or apoptotic response that is the ultimate goal of molecularly targeted agents discussed above. However, there is much commonality in the strategies to evaluate and use molecularly targeted and biologic therapies.
Immune Cell–Mediated Therapies Tumors have a variety of means of avoiding the immune system: (1) they are often only subtly different from their normal counterparts; (2) they are capable of downregulating their major histocompatibility complex antigens, effectively masking them from recognition by T cells; (3) they are inefficient at presenting antigens to the immune system; (4) they can cloak themselves in a protective shell of fibrin to minimize contact with surveillance mechanisms; and (5) they can produce a range of soluble molecules, including potential immune targets, that can distract the immune system from recognizing the tumor cell or can kill or inactivate the immune effector cells. Some of the cell products initially polarize the immune response away from cellular immunity (shifting from TH1 to TH2 responses; Chap. 372e) and ultimately lead to defects in T cells that prevent their activation and cytotoxic activity. Cancer treatment further suppresses host immunity. A variety of strategies are being tested to overcome these barriers.
Cell-Mediated Immunity The strongest evidence that the immune system can exert clinically meaningful antitumor effects comes from allogeneic bone marrow transplantation. Adoptively transferred T cells from the donor expand in the tumor-bearing host, recognize the tumor as being foreign, and can mediate impressive antitumor effects (graft-versus-tumor effects). Three types of experimental interventions are being developed to take advantage of the ability of T cells to kill tumor cells.
1. Transfer of allogeneic T cells. This occurs in three major settings: in allogeneic bone marrow transplantation; as purified lymphocyte transfusions following bone marrow recovery after allogeneic bone marrow transplantation; and as pure lymphocyte transfusions following immunosuppressive (nonmyeloablative) therapy (also called minitransplants). In each of these settings, the effector cells are donor T cells that recognize the tumor as being foreign, probably through minor histocompatibility differences. The main risk of such therapy is the development of graft-versus-host disease because of the minimal difference between the cancer and the normal host cells. This approach has been highly effective in certain hematologic cancers.
2. Transfer of autologous T cells. In this approach, the patient’s own T cells are removed from the tumor-bearing host, manipulated in several ways in vitro, and given back to the patient. There are three major classes of autologous T-cell manipulation. First, tumor antigen–specific T cells can be developed and expanded to large numbers over many weeks ex vivo before administration. Second, the patient’s T cells can be activated by exposure to polyclonal stimulators such as anti-CD3 and anti-CD28 after a short period ex vivo, and then amplified in the host after transfer by stimulation with IL-2, for example. Short periods removed from the patient permit the cells to overcome the tumor-induced T cell defects, and such cells traffic and home to sites of disease better than cells that have been in culture for many weeks. In a third approach, genes that encode for a T cell receptor specific for an antigen expressed by the tumor along with genes that facilitate T cell activation can be introduced into subsets of a patient’s T cells, which, after transfer back into the patient, allow homing of cytotoxic T cells to tumor cells expressing the antigen.
3. Tumor vaccines aimed at boosting T cell immunity. The finding that mutant oncogenes that are expressed only intracellularly can be recognized as targets of T cell killing greatly expanded the possibilities for tumor vaccine development. No longer is it difficult to find something different about tumor cells. However, major difficulties remain in getting the tumor-specific peptides presented in a fashion to prime the T cells. Tumors themselves are very poor at presenting their own antigens to T cells at the first antigen exposure (priming). Priming is best accomplished by professional antigen-presenting cells (dendritic cells). Thus, a number of experimental strategies are aimed at priming host T cells against tumor-associated peptides. Vaccine adjuvants such as granulocyte-macrophage colony-stimulating factor (GM-CSF) appear capable of attracting antigen-presenting cells to a skin site containing a tumor antigen. Such an approach has been documented to eradicate microscopic residual disease in follicular lymphoma and give rise to tumor-specific T cells. Purified antigen-presenting cells can be pulsed with tumor, its membranes, or particular tumor antigens and delivered as a vaccine. One such vaccine, Sipuleucel-T, is approved for use in patients with hormone-independent prostate cancer. In this approach, the patient undergoes leukapheresis, wherein mononuclear cells (that include antigen-presenting cells) are removed from the patient’s blood. The cells are pulsed in a laboratory with an antigenic fusion protein comprising a protein frequently expressed by prostate cancer cells, prostate acid phosphatase, fused to GM-CSF, and matured to increase their capacity to present the antigen to immune effector cells. The cells are then returned to the patient in a well-tolerated treatment. Although no objective tumor response was documented in clinical trials, median survival was increased by about 4 months. Tumor cells can also be transfected with genes that attract antigen-presenting cells.
Another important vaccine strategy is directed at infectious agents whose action ultimately is tied to the development of human cancer. Hepatitis B vaccine in an epidemiologic sense prevents hepatocellular carcinoma, and a tetravalent human papillomavirus vaccine prevents infection by virus types currently accounting for 70% of cervical cancer. Unfortunately, these vaccines are ineffective at treating patients who have developed a virus-induced cancer.
Antibody-Mediated Therapeutic Approaches In general, antibodies are not very effective at killing cancer cells. Because the tumor seems to influence the host toward making antibodies rather than generating cellular immunity, it is inferred that antibodies are easier for the tumor to fend off. Many patients can be shown to have serum antibodies directed at their tumors, but these do not appear to influence disease progression. However, the ability to grow very large quantities of high-affinity antibody directed at a tumor by the hybridoma technique has led to the application of antibodies in the treatment of cancer. In this approach, antibodies are derived where the antigen-combining regions are grafted onto human immunoglobulin gene products (chimerized or humanized) or derived de novo from mice bearing human immunoglobulin gene loci. Three general strategies have emerged using antibodies. Tumor-regulatory antibodies target tumor cells directly or indirectly to modulate intracellular functions or attract immune or stromal cells. Immunoregulatory antibodies target antigens expressed on the tumor cells or host immune cells to modulate primarily the host’s immune responsiveness to the tumor. Finally, antibody conjugates can be made with the antibody linked to drugs, toxins, or radioisotopes to target these “warheads” for delivery to the tumor. Table 103e-6 lists features of currently used or promising antibodies for cancer treatment.
ANTIBODIES USED IN CANCER TREATMENT |
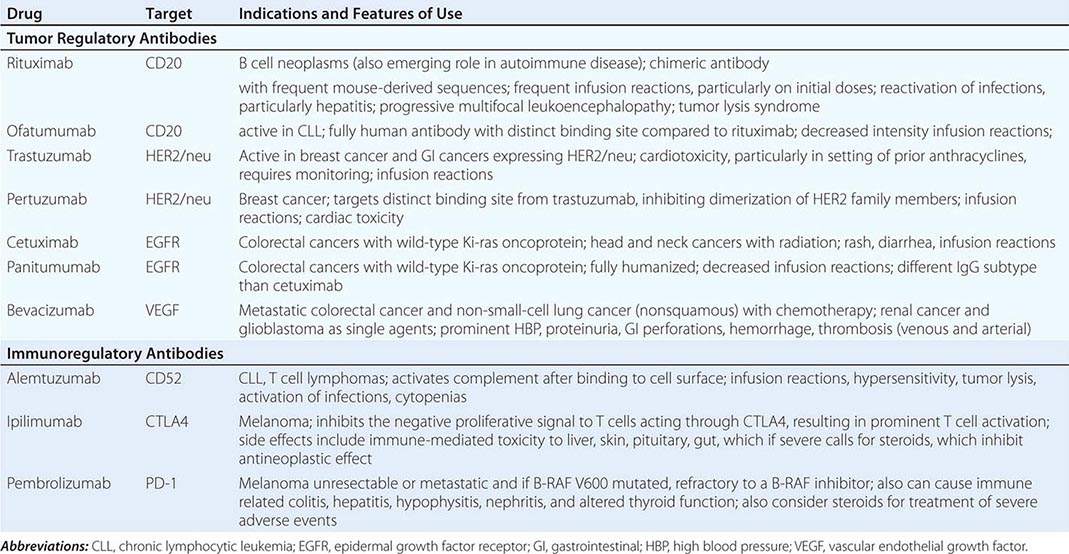
TUMOR-REGULATORY ANTIBODIES Humanized antibodies against the CD20 molecule expressed on B cell lymphomas (rituximab and ofatumumab) are exemplary of antibodies that affect both signaling events driving lymphomagenesis as well as activating immune responses against B cell neoplasms. They are used as single agents and in combination with chemotherapy and radiation in the treatment of B cell neoplasms. Obinutuzumab is an antibody with an altered glycosylation that enhances its ability to fix complement; it is also directed against CD20 and is of value in chronic lymphocytic leukemia. It seems to be more effective in this setting than rituximab.
The HER2/neu receptor overexpressed on epithelial cancers, especially breast cancer, was initially targeted by trastuzumab, with noteworthy activity in potentiating the action of chemotherapy in breast cancer as well as some evidence of single-agent activity. Trastuzumab also appears to interrupt intracellular signals derived from HER2/neu and to stimulate immune mechanisms. The anti-HER2 antibody pertuzumab, specifically targeting the domain of HER2/neu responsible for dimerization with other HER2 family members, is more specifically directed against HER2 signaling function and augments the action of trastuzumab.
EGF receptor (EGFR)-directed antibodies (such as cetuximab and panitumumab) have activity in colorectal cancer refractory to chemotherapy, particularly when used to augment the activity of an additional chemotherapy program, and in the primary treatment of head and neck cancers treated with radiation therapy. The mechanism of action is unclear. Direct effects on the tumor may mediate an antiproliferative effect as well as stimulate the participation of host mechanisms involving immune cell or complement-mediated response to tumor cell–bound antibody. Alternatively, the antibody may alter the release of paracrine factors promoting tumor cell survival.
The anti-VEGF antibody bevacizumab shows little evidence of antitumor effect when used alone, but when combined with chemotherapeutic agents, it improves the magnitude of tumor shrinkage and time to disease progression in colorectal and nonsquamous lung cancers. The mechanism for the effect is unclear and may relate to the capacity of the antibody to alter delivery and tumor uptake of the active chemotherapeutic agent. Ziv-aflibercept is not an antibody, but a solubilized VEGF receptor VEGF binding domain, and therefore may have a distinct mechanism of action with comparable side effects.
Unintended side effects of any antibody use include infusion-related hypersensitivity reactions, usually limited to the first infusion, which can be managed with glucocorticoid and/or antihistamine prophylaxis. In addition, distinct syndromes have emerged with different antibodies. Anti-EGFR antibodies produce an acneiform rash that poorly responds to glucocorticoid cream treatment. Trastuzumab (anti-HER2) can inhibit cardiac function, particularly in patients with prior exposure to anthracyclines. Bevacizumab has a number of side effects of medical significance, including hypertension, thrombosis, proteinuria, hemorrhage, and gastrointestinal perforations with or without prior surgeries; these adverse events also occur with small-molecule drugs modulating VEGFR function.
IMMUNOREGULATORY ANTIBODIES Purely immunoregulatory antibodies stimulate immune responses to mediate tumor-directed cytotoxicity. First-generation approaches sought to activate complement and are exemplified by antibodies to CD52; these are active in chronic lymphoid leukemia and T cell malignancies. A more refined understanding of the tumor–host interface has defined that cytotoxic tumor-directed T cells are frequently inhibited by ligands upregulated in the tumor cells. The programmed death ligand 1 (PD-L1; also known as B7-homolog 1) was initially recognized as an entity that induced T cell death through a receptor present on T cells, termed the PD receptor (Fig 103e-5), which physiologically exists to regulate the intensity of the immune response. The PD family of ligands and receptors also regulates macrophage function, present in tumor stroma. These actions raised the hypothesis that antibodies directed against the PD signaling axis (both anti-PD-L1 and anti-PD) might be useful in cancer treatment by allowing reactivation of the immune response against tumors. Indeed, nivolumab and lambrolizumab, both anti-PD antibodies, have shown evidence of important immune-mediated actions against certain solid tumors, including melanoma and lung cancers.
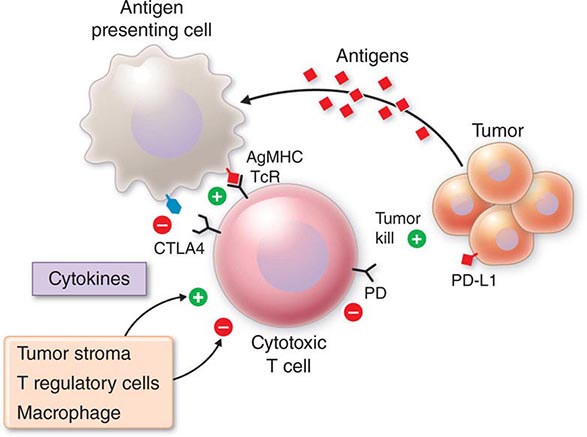
FIGURE 103e-5 Tumors possess a microenvironment (tumor stroma) with immune cells including both helper T cells, suppressor T cells (both “regulatory” of other immune cell function), macrophages, and cytotoxic T cells. Cytokines found in the stroma and deriving from macrophawsges and regulatory T cells modulate the activities of cytotoxic T cells, which have the potential to kill tumor cells. Antigens released by tumor cells are taken up by Antigen Presenting Cells (APCs), also in the stroma. Antigens are processed by the APCs to peptides presented by the Major Histocompatibility Complex to T-cell antigen receptors, thus providing an (+) activation signal for the cytotoxic tumor cells to kill tumor cells bearing that antigen. Negative (–) signals inhibiting cytoxic T cell action include the CTLA4 receptor (on T cells), interacting with the B7 family of negative regulatory signals from APCs, and the PD receptor (on T cells), interacting with the PD-L1 (–) signal coming from tumor cells expressing the PD-1 ligand (PD-l1). As both CTLA4 and PD1 signals attenuate the anti-tumor T cell response, strategies which inhibit CTLA4 and PD1 function are a means of stimulating cytotoxic T cell activity to kill tumor cells. Cytokines from other immune cells and macrophages can provide both (+) and (–) signals for T cell action, and are under investigation as novel immunoregulatory therapeutics.
Already approved for clinical use in melanoma is ipilimumab, an antibody directed against the anti-CTLA4 (cytotoxic T lymphocyte antigen 4), which is expressed on T cells (not tumor cells), responds to signals from antigen-presenting cells (Fig. 103e-5), and also downregulates the intensity of the T cell proliferative response to antigens derived from tumor cells. Indeed, manipulation of the CTLA4 axis was the first demonstration that purely immunoregulatory antibody strategies directed at T cell physiology could be safe and effective in the treatment of cancer, although it acts at a very early stage in T cell activation and can be considered somewhat nonspecific in its basis for T cell stimulation. Pembrolizumab, an anti-PD ligand blocking agent was also approved for melanoma, with a similar spectrum of potential adverse events, but acting in the tumor microenvironment. Indeed, prominent activation of autoimmune hepatic, endocrine, cutaneous, neurologic, and gastrointestinal responses is a basis for adverse events with the use of ipilimumab; the emergent use of glucocorticoids may be required to attenuate severe toxicities, which unfortunately can cause potential attenuation of antitumor effect. Importantly for the general internist, these events may occur late after exposure to ipilimumab while the patient may otherwise be enjoying sustained control of tumor growth owing to the beneficial actions of ipilimumab.
Another class of immunoregulatory antibody is the “bispecific” antibody blinatumomab, which was constructed to have an anti-CD19 antigen combining site as one valency of an antibody with anti-CD3 binding site as the other valency. This antibody thus can bring T cells (with its anti-CD3 activity) close to B cells bearing the CD19 determinant. Blinatumomab is active in B cell neoplasms such as acute lymphocytic leukemia, which may not have prominent expression of the CD20 targeted by rituximab.
ANTIBODY CONJUGATES Conjugates of antibodies with drugs and isotopes have also been shown to be effective in the treatment of cancer and have the intent of increasing the therapeutic index of the drug or isotope by delivering the toxic “warhead” directly to the tumor cell or tumor microenvironment. Ado-trastuzumab is a conjugate of the HER2/neu-directed trastuzumab and a highly toxic microtubule targeted drug (emtansine), which by itself is too toxic for human use; the antibody-drug conjugate shows valuable activity in patients with breast cancer who have developed resistance to the “naked” antibody. Brentuximab vedotin is an anti-CD30 antibody drug conjugate with a distinct microtubule poison with activity in neoplasms such as Hodgkin’s lymphoma where the tumor cells frequently express CD30. Radioconjugates targeting CD20 on lymphomas have been approved for use (ibritumomab tiuxetan [Zevalin], using yttrium-90 or 131I-tositumomab). Toxicity concerns have limited their use.
Cytokines There are >70 separate proteins and glycoproteins with biologic effects in humans: interferon (IFN) α, β, and γ; interleukin (IL) 1 through 29 (so far); the tumor necrosis factor (TNF) family (including lymphotoxin, TNF-related apoptosis-inducing ligand [TRAIL], CD40 ligand, and others); and the chemokine family. Only a fraction of these has been tested against cancer; only IFN-α and IL-2 are in routine clinical use.
About 20 different genes encode IFN-α, and their biologic effects are indistinguishable. IFN induces the expression of many genes, inhibits protein synthesis, and exerts a number of different effects on diverse cellular processes. The two recombinant forms that are commercially available are IFN-α2a and -α2b. Interferon is not curative for any tumor but can induce partial responses in follicular lymphoma, hairy cell leukemia, CML, melanoma, and Kaposi’s sarcoma. It has been used in the adjuvant setting in stage II melanoma, multiple myeloma, and follicular lymphoma, with uncertain effects on survival. It produces fever, fatigue, a flulike syndrome, malaise, myelosuppression, and depression and can induce clinically significant autoimmune disease. IFN-α is not generally the treatment of choice for any cancer.
IL-2 exerts its antitumor effects indirectly through augmentation of immune function. Its biologic activity is to promote the growth and activity of T cells and natural killer (NK) cells. High doses of IL-2 can produce tumor regression in certain patients with metastatic melanoma and renal cell cancer. About 2–5% of patients may experience complete remissions that are durable, unlike any other treatment for these tumors. IL-2 is associated with myriad clinical side effects, including intravascular volume depletion, capillary leak syndrome, adult respiratory distress syndrome, hypotension, fever, chills, skin rash, and impaired renal and liver function. Patients may require blood pressure support and intensive care to manage the toxicity. However, once the agent is stopped, most of the toxicities reverse completely within 3–6 days.
Ligand Receptor–Directed Constructs High-affinity receptors for cytokines have led to the design of cytokine-toxin recombinant fusion proteins, such as IL-2 expressed in frame with a fragment of diphtheria toxin. A commercially available construct has activity against certain T cell lymphomas. Likewise, the high-affinity folate receptor is the target for folate conjugated to chemotherapeutic agents. In both cases, the drug’s utility derives from the internalization of the targeted receptor and cleavage of the active drug or toxin moiety.
SYSTEMIC RADIATION THERAPY
Although total-body irradiation has a role in preparing a patient to received allogeneic stem cells, and antibodies as described above can specifically target radioisotopes, systemically administered isotopes of iodide salts have an important role in the treatment of thyroid neoplasms, owing to the selective upregulation of the iodide transporter in the tumor cell compartment. Likewise, isotopes of samarium and radium have been found useful in the palliation of symptoms from advanced bony metastases of prostate cancer owing to their selective deposition at the tumor–bone matrix interface, thereby potentially affecting the function of both tumor and stromal cells in the progressive growth of the metastatic deposit.
RESISTANCE TO CANCER TREATMENTS
Resistance mechanisms to the conventional cytotoxic agents were initially characterized in the late twentieth century as defects in drug uptake, metabolism, or export by tumor cells. The multidrug resistance (mdr) gene defined in vitro in cell lines exposed to increasing concentrations of drugs led to the definition of a family of transport proteins that, when overexpressed, result in the facile transport of a variety of hydrophobic drugs out of the cancer cell. Although efforts to manipulate this transporter to promote drug residence in tumor cells have been pursued, none are clinically useful at this time. Drug-metabolizing enzymes such as cytidine deaminase are upregulated in resistant tumor cells, and this is the basis for so-called “high-dose cytarabine” regimens in the treatment of leukemia. Another resistance mechanism defined during this era involved increased expression of a drug’s target, exemplified by amplification of the dihydrofolate reductase gene, in patients who had lost responsiveness to methotrexate, or mutation of topoisomerase II in tumors that relapsed after topoisomerase II modulator treatment.
A second class of resistance mechanisms involves loss of the cellular apoptotic mechanism activated after the engagement of a drug’s target by the drug. This occurs in a way that is heavily influenced by the biology of the particular tumor type. For example, decreased alkylguanine alkyltransferase defines a subset of glioblastoma patients with the prospect of greatest benefit from treatment with temozolomide, but has no predictive value for benefit from temozolomide in epithelial neoplasms. Likewise, ovarian cancers resistant to platinating agents have decreased expression of the proapoptotic gene bax. These types of findings have prompted the idea that responsive tumors to chemotherapeutic agents are populated by cells that express drug-related cell death controlling genes, creating in effect a state of “synthetic lethality” of the drug (Chap. 102e) with the genes expressed in responsive tumors, analogous to the existence in yeast of mutations that are well tolerated in the absence of a physiologic stressor but become lethal in the presence of that stressor. In the case of tumors, the chemotherapy inducing the cell death response is the analogous physiologic stressor.
A third class of resistance mechanisms emerged from sequencing of the targets of agents directed at oncogenic kinases. Thus, patients with CML resistant to imatinib have acquired mutations in the ATP binding domain of p210bcr-abl in some cases, leading to the screening and design of agents with activity against the mutant proteins. Entirely analogous resistance mechanisms have emerged in patients with lung cancer treated with the EGFR antagonists gefitinib and erlotinib.
A final category of tumor resistance mechanisms to targeted agents includes the upregulation of alternate means of activating the pathway targeted by the agent. Thus melanomas initially responsive to BRAF V600E antagonists such as vemurafenib may reactivate raf signaling by upregulating isoforms that can bypass the variant blocked by the drug. Likewise, inhibition of HER2/neu signaling in breast cancer cells can lead to the emergence of variants with distinct oncogenic signaling pathways such as PI3 kinase. Analogously in NSCLC, EGFR inhibitor treatment leads to the emergence of cells with a predominance of c-met protooncogene–dependent signaling in the resistant tumors.
The susceptibility of a tumor to different treatments as a function of its expression of potential drug targets or their mutational profile has led to efforts to define the dominant pathways driving a patient’s tumor by genomic techniques including whole exome sequencing. The difficulty with applying such data to patient treatment is recognizing that these pathways may change during the natural history of a tumor and that different sites in a single patient may have tumors with different patterns of gene mutation.
SUPPORTIVE CARE DURING CANCER TREATMENT
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


