Immunosuppressants, Tolerogens, and Immunostimulants
This chapter reviews the components of the immune response and drugs that modulate immunity in 3 ways: immunosuppression, tolerance, and immunostimulation. Four major classes of immunosuppressive drugs are discussed: glucocorticoids (see Chapter 42), calcineurin inhibitors, antiproliferative and antimetabolic agents (see Chapter 61), and antibodies. The chapter ends with a brief case study of immunotherapy for MS.
THE IMMUNE RESPONSE
The immune system evolved to discriminate self from nonself. Innate immunity (natural immunity) is primitive, does not require priming, and is of relatively low affinity, but is broadly reactive. Adaptive immunity (learned immunity) is antigen specific, depends on antigen exposure or priming, and can be of very high affinity. The 2 arms of immunity work closely together, with the innate immune system being most active early in an immune response and adaptive immunity becoming progressively dominant over time.
The major effectors of innate immunity are complement, granulocytes, monocytes/macrophages, natural killer cells, mast cells, and basophils. The major effectors of adaptive immunity are B and T lymphocytes. B lymphocytes make antibodies; T lymphocytes function as helper, cytolytic, and regulatory (suppressor) cells. These cells are important in the normal immune response to infection and tumors but also mediate transplant rejection and autoimmunity.
Immunoglobulins (antibodies) on the B-lymphocyte surface are receptors for a large variety of specific structural conformations. In contrast, T lymphocytes recognize antigens as peptide fragments in the context of self major histocompatibility complex (MHC) antigens (called human leukocyte antigens [HLAs] in humans) on the surface of antigen-presenting cells, such as dendritic cells, macrophages, and other cell types expressing MHC class I and class II antigens. Once activated by specific antigen recognition, both B and T lymphocytes are triggered to differentiate and divide, leading to release of soluble mediators (cytokines, lymphokines) that perform as effectors and regulators of the immune response.
IMMUNOSUPPRESSION
Immunosuppressive drugs are used to dampen the immune response in organ transplantation and autoimmune disease. In transplantation, the major classes of immunosuppressive drugs used today are:
• Glucocorticoids
• Calcineurin inhibitors
• Antiproliferative/antimetabolic agents
• Biologicals (antibodies)
Table 35–1 summarizes the sites of action of representative immunosuppressants on T-cell activation.
Table 35–1
Sites of Action of Selected Immunosuppressive Agents on T-Cell Activation

These drugs are used in treating conditions such as acute immune rejection of organ transplants and severe auto-immune diseases. However, such therapies require lifelong use and nonspecifically suppress the entire immune system, exposing patients to considerably higher risks of infection and cancer. The calcineurin inhibitors and glucocorticoids, in particular, are nephrotoxic and diabetogenic, respectively, thus restricting their usefulness in a variety of clinical settings. Monoclonal and polyclonal antibody preparations directed at reactive T cells are important adjunct therapies and provide a unique opportunity to target specifically immune-reactive cells. Finally, newer small molecules and antibodies have expanded the arsenal of immunosuppressives. In particular, mammalian target of rapamycin (mTOR) inhibitors (sirolimus, everolimus) and anti-CD25 (interleukin-2 receptor [IL-2R]) antibodies (basiliximab, daclizumab) target growth-factor pathways, substantially limiting clonal expansion and thus potentially promoting tolerance.
GENERAL APPROACH TO ORGAN TRANSPLANTATION THERAPY. Organ transplantation therapy is organized around 5 general principles.
• Carefully prepare patient and select the best available ABO blood type–compatible HLA match for organ donation.
• Employ multitiered immunosuppressive therapy; simultaneously use several agents, each of which is directed at a different molecular target within the allograft response. Synergistic effects permit use of the various agents at relatively low doses, thereby limiting specific toxicities while maximizing the immunosuppressive effect.
• Employ intensive induction and lower-dose maintenance drug protocols; greater immunosuppression is required to gain early engraftment and/or to treat established rejection than to maintain long-term immunosuppression.
• Investigation of each episode of transplant dysfunction is required, including evaluation for rejection, drug toxicity, and infection.
• Reduce dosage or withdraw a drug if its toxicity exceeds its benefit.
BIOLOGICAL INDUCTION THERAPY. Induction therapy with polyclonal and monoclonal antibodies (mAbs) has been an important component of immunosuppression since the 1960s, when Starzl and colleagues demonstrated the beneficial effect of antilymphocyte globulin (ALG) in the prophylaxis of rejection. Two preparations are FDA-approved for use in transplantation: lymphocyte immune globulin (ATGAM) and antithymocyte globulin (ATG; THYMOGLOBULIN). ATG is the most frequently used depleting agent. Alemtuzumab, a humanized anti-CD52 monoclonal antibody that produces prolonged lymphocyte depletion, is approved for use in chronic lymphocytic leukemia but is increasingly used off label as induction therapy in transplantation.
In many transplant centers, induction therapy with biological agents is used to delay the use of the nephrotoxic calcineurin inhibitors or to intensify the initial immunosuppressive therapy in patients at high risk of rejection (i.e., repeat transplants, broadly presensitized patients, African American patients, or pediatric patients). Most of the limitations of murine-based mAbs generally were overcome by the introduction of chimeric or humanized mAbs that lack antigenicity and have a prolonged serum t1/2. Antibodies derived from transgenic mice carrying human antibody genes are labeled “humanized” (90-95% human) or “fully human” (100% human); antibodies derived from human cells are labeled “human.” However, all 3 types of antibodies probably are of equal efficacy and safety. Chimeric antibodies generally contain ~33% mouse protein and 67% human protein and can still produce an antibody response, resulting in reduced efficacy and shorter t1/2 compared to humanized antibodies.
Biological agents for induction therapy in the prophylaxis of rejection currently are used in ~70% of de novo transplant patients. Biological agents for induction can be divided into 2 groups: the depleting agents and the immune modulators. The depleting agents consist of lymphocyte immune globulin, ATG, and muromonab-CD3 mAb; their efficacy derives from their ability to deplete the recipient’s CD3-positive cells at the time of transplantation and antigen presentation. The second group of biological agents, the anti–IL-2R mAbs, do not deplete T lymphocytes, with the possible exception of T regulatory cells, but rather block IL-2–mediated T-cell activation by binding to the α chain of IL-2R. For patients with high levels of anti-HLA antibodies and humoral rejection, more aggressive therapies include plasmapheresis, intravenous immunoglobulin, and rituximab, a chimeric anti-CD20 monoclonal antibody.
MAINTENANCE IMMUNOTHERAPY. Basic immunosuppressive therapy uses multiple drugs simultaneously, typically a calcineurin inhibitor, glucocorticoids, and mycophenolate (a purine metabolism inhibitor), each directed at a discrete site in T-cell activation. Glucocorticoids, azathioprine, cyclosporine, tacrolimus, mycophenolate, sirolimus, and various monoclonal and polyclonal antibodies all are approved for use in transplantation.
THERAPY FOR ESTABLISHED REJECTION. Low doses of prednisone, calcineurin inhibitors, purine metabolism inhibitors, or sirolimus are effective in preventing acute cellular rejection; they are less effective in blocking activated T lymphocytes and thus are not very effective against established, acute rejection or for the total prevention of chronic rejection. Therefore, treatment of established rejection requires the use of agents directed against activated T cells. These include glucocorticoids in high doses (pulse therapy), polyclonal antilymphocyte antibodies, or muromonab-CD3.
GLUCOCORTICOIDS
The glucocorticoids are described in Chapter 42. Prednisone, prednisolone, and other glucocorticoids are used alone and in combination with other immunosuppressive agents for treatment of transplant rejection and autoimmune disorders.
Mechanism of Action. Glucocorticoids have broad anti-inflammatory effects on multiple components of cellular immunity, but relatively little effect on humoral immunity. Glucocorticoids bind to receptors inside cells and regulate the transcription of numerous other genes (see Chapter 42). Glucocorticoids also curtail activation of NF-κB, suppress formation of pro-inflammatory cytokines such as IL-1 and IL-6, inhibit T cells from making IL-2 and proliferating, and inhibit the activation of cytotoxic T lymphocytes. In addition, neutrophils and monocytes display poor chemotaxis and decreased lysosomal enzyme release.
Therapeutic Uses. Glucocorticoids commonly are combined with other immunosuppressive agents to prevent and treat transplant rejection. Glucocorticoids also are efficacious for treatment of graft-versus-host disease in bone-marrow transplantation. Glucocorticoids are routinely used to treat autoimmune disorders such as rheumatoid and other arthritides, systemic lupus erythematosus, systemic dermatomyositis, psoriasis and other skin conditions, asthma and other allergic disorders, inflammatory bowel disease, inflammatory ophthalmic diseases, auto-immune hematological disorders, and acute exacerbations of MS (see “Multiple Sclerosis,” below). In addition, glucocorticoids limit allergic reactions that occur with other immunosuppressive agents and are used in transplant recipients to block first-dose cytokine storm caused by treatment with muromonab-CD3 and to a lesser extent ATG (see “Antithymocyte Globulin”).
Toxicity. Extensive steroid use often results in disabling and life-threatening adverse effects described in Chapter 42. The advent of combined glucocorticoid/calcineurin inhibitor regimens has allowed reduced doses or rapid withdrawal of steroids, resulting in lower steroid-induced morbidities.
CALCINEURIN INHIBITORS
The most effective immunosuppressive drugs in routine use are the calcineurin inhibitors, cyclosporine and tacrolimus, which target intracellular signaling pathways induced as a consequence of T-cell–receptor activation (Figure 35–1). Cyclosporine and tacrolimus bind to an immunophilin (cyclophilin for cyclosporine, or FKBP-12 for tacrolimus), resulting in subsequent interaction with calcineurin to block its phosphatase activity. Calcineurin-catalyzed dephosphorylation is required for movement of a component of the nuclear factor of activated T lymphocytes (NFAT) into the nucleus. NFAT, in turn, is required to induce a number of cytokine genes, including that for IL-2, a prototypic T-cell growth and differentiation factor.
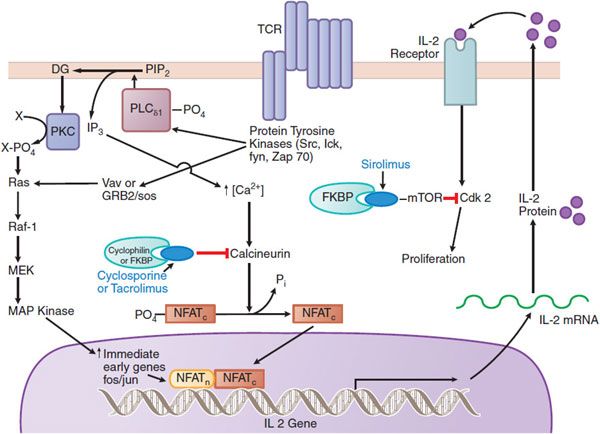
Figure 35–1 Mechanisms of action of cyclosporine, tacrolimus, and sirolimus on T cells. Cyclosporine and tacrolimus bind to immunophilins (cyclophilin and FK506-binding protein [FKBP], respectively), forming a complex that inhibits the phosphatase calcineurin and the calcineurin-catalyzed dephosphorylation that permits translocation of nuclear factor of activated T cells (NFAT) into the nucleus. NFAT is required for transcription of interleukin-2 (IL-2) and other growth and differentiation–associated cytokines (lymphokines). Sirolimus (rapamycin) works downstream of the IL-2 receptor, binding to FKBP; the FKBP-sirolimus complex binds to and inhibits the mammalian target of rapamycin (mTOR), a kinase involved in cell-cycle progression (proliferation). TCR, the T-cell receptor that recognizes antigens bound to the major histocompatibility complex. (Reproduced with permission from Clayberger C, Krensky AM. Mechanisms of allograft rejection. In Neilson EG, Couser WG, eds, Immunologic Renal Diseases, 2nd ed. Philadelphia: Lippincott Williams & Wilkins, 2001, pp 321–346. http://lww.com.)
TACROLIMUS. Tacrolimus (PROGRAF, FK506) is a macrolide antibiotic produced by Streptomyces tsukubaensis. Because of perceived slightly greater efficacy and ease of blood level monitoring, tacrolimus has become the preferred calcineurin inhibitor in most transplant centers.
Mechanism of Action. Like cyclosporine, tacrolimus inhibits T-cell activation by inhibiting calcineurin. Tacrolimus binds to an intracellular protein, FK506-binding protein–12 (FKBP-12), an immunophilin structurally related to cyclophilin. A complex of tacrolimus-FKBP-12, Ca2+, calmodulin, and calcineurin then forms, and calcineurin phosphatase activity is inhibited (see Figure 35–1). Inhibition of phosphatase activity prevents dephosphorylation and nuclear translocation of NFAT and inhibits T-cell activation. Thus, although the intracellular receptors differ, cyclosporine and tacrolimus target the same pathway for immunosuppression.
ADME. Tacrolimus is available for oral administration as capsules (0.5, 1, and 5 mg) and as a solution for injection (5 mg/mL). Because of intersubject variability in pharmacokinetics, individualized dosing is required for optimal therapy. For tacrolimus, whole blood seems to be the best sampling compartment; the trough drug level in whole blood seems to correlate better with clinical events for tacrolimus than for cyclosporine. Target concentrations are 10-15 ng/mL in the early preoperative period and 100-200 ng/mL 3 months after transplantation. GI absorption is incomplete and variable. Food decreases the rate and extent of absorption. Plasma protein binding of tacrolimus is 75-99%, involving primarily albumin and α1-acid glycoprotein. The t1/2 of tacrolimus is ~12 h. Tacrolimus is extensively metabolized in the liver by CYP3A; some of the metabolites are active. The bulk of excretion of the parent drug and metabolites is in the feces.
Therapeutic Uses. Tacrolimus is indicated for the prophylaxis of solid-organ allograft rejection in a manner similar to cyclosporine (see “Cyclosporine”) and is used off label as rescue therapy in patients with rejection episodes despite “therapeutic” levels of cyclosporine. Recommended initial oral doses are 0.2 mg/kg/day for adult kidney transplant patients, 0.1-0.15 mg/kg/day for adult liver transplant patients, 0.075 mg/kg/day for adult heart transplant patients, and 0.15-0.2 mg/kg/day for pediatric liver transplant patients in 2 divided doses 12 h apart. These dosages are intended to achieve typical blood trough levels in the 5- to 20-ng/mL range.
Toxicity. Nephrotoxicity, neurotoxicity (e.g., tremor, headache, motor disturbances, seizures), GI complaints, hypertension, hyperkalemia, hyperglycemia, and diabetes all are associated with tacrolimus use. Tacrolimus has a negative effect on pancreatic islet β cells, and glucose intolerance and diabetes mellitus are well-recognized complications of tacrolimus-based immunosuppression. As with other immunosuppressive agents, there is an increased risk of secondary tumors and opportunistic infections. Notably, tacrolimus does not adversely affect uric acid or LDL cholesterol. Diarrhea and alopecia are common in patients on concomitant mycophenolate therapy.
Drug Interactions. Because of its potential for nephrotoxicity, tacrolimus blood levels and renal function should be monitored closely. Coadministration with cyclosporine results in additive or synergistic nephrotoxicity; therefore, a delay of at least 24 h is required when switching a patient from cyclosporine to tacrolimus. Because tacrolimus is metabolized mainly by CYP3A, the potential interactions described in the following section for cyclosporine also apply for tacrolimus.
CYCLOSPORINE. Cyclosporine (cyclosporin A), a cyclic polypeptide of 11 amino acids, is produced by the fungus Beauveria nivea. Figure 35–1 depicts the molecular action of cyclosporine to inhibit calcineurin activity. At the level of immune system function, cyclosporine suppresses some humoral immunity but is more effective against T cell–dependent immune mechanisms such as those underlying transplant rejection and some forms of autoimmunity. It preferentially inhibits antigen-triggered signal transduction in T lymphocytes, blunting expression of many lymphokines, including IL-2, and the expression of anti-apoptotic proteins. Cyclosporine also increases expression of transforming growth factor β (TGF-β), a potent inhibitor of IL-2–stimulated T-cell proliferation and generation of cytotoxic T lymphocytes (CTLs).
ADME. Because cyclosporine is lipophilic and highly hydrophobic, it is formulated for clinical administration using castor oil or other strategies to ensure solubilization. Cyclosporine can be administered intravenously or orally. The intravenous preparation (SANDIMMUNE, others) is provided as a solution in an ethanol-polyoxyethylated castor oil vehicle that must be further diluted in 0.9% sodium chloride solution or 5% dextrose solution before injection. The oral dosage forms include soft gelatin capsules and oral solutions. Cyclosporine supplied in the original soft gelatin capsule is absorbed slowly, with 20-50% bioavailability. A modified microemulsion formulation (NEORAL) has become the most widely used preparation. It has more uniform and slightly increased bioavailability compared to the original formulation. It is provided as 25-mg and 100-mg soft gelatin capsules and a 100-mg/mL oral solution. The original and microemulsion formulations are not bioequivalent and cannot be used interchangeably without supervision by a physician and monitoring of drug concentrations in plasma. Generic preparations of both NEORAL and SANDIMMUNE are bioequivalent by FDA criteria. Transplant units need to educate patients that SANDIMMUNE and its generics are not the same as NEORAL and its generics, such that one preparation cannot be substituted for another without risk of inadequate immunosuppression or increased toxicity.
Blood levels taken 2 h after a dose administration (so-called C2 levels) may correlate better with the AUC than other single points, but no single time point can simulate the exposure better than more frequent drug sampling. In practice, if a patient has clinical signs or symptoms of toxicity, or if there is unexplained rejection or renal dysfunction, a pharmacokinetic profile can be used to estimate that person’s exposure to the drug.
Cyclosporine absorption is incomplete following oral administration and varies with the individual patient and the formulation used. The elimination of cyclosporine from the blood generally is biphasic, with a terminal t1/2 of 5-18 h. After intravenous infusion, clearance is ~5-7 mL/min/kg in adult recipients of renal transplants, but results differ by age and patient populations. For example, clearance is slower in cardiac transplant patients and more rapid in children. Thus, the intersubject variability is so large that individual monitoring is required. After oral administration of cyclosporine (as NEORAL), the time to peak blood concentrations is 1.5-2 h. Administration with food delays and decreases absorption. High- and low-fat meals consumed within 30 min of administration decrease the AUC by ~13% and the maximum concentration by 33%. This makes it imperative to individualize dosage regimens for outpatients. Cyclosporine is extensively metabolized by hepatic CYP3A and to a lesser degree by the GI tract and kidneys. Cyclosporine and its metabolites are excreted principally through the bile into the feces, with ~6% excreted in the urine. Cyclosporine also is excreted in human milk. In the presence of hepatic dysfunction, dosage adjustments are required. No adjustments generally are necessary for dialysis or renal failure patients.
Therapeutic Uses. Clinical indications for cyclosporine are kidney, liver, heart, and other organ transplantation; rheumatoid arthritis; and psoriasis. Its use in dermatology is discussed in Chapter 65. Cyclosporine usually is combined with other agents, especially glucocorticoids and either azathioprine or mycophenolate and, most recently, sirolimus. The dose of cyclosporine varies, depending on the organ transplanted and the other drugs used in the specific treatment protocol(s). The initial dose generally is not given before the transplant because of the concern about nephrotoxicity. Dosing is guided by signs of rejection (too low a dose), renal or other toxicity (too high a dose), and close monitoring of blood levels. Great care must be taken to differentiate renal toxicity from rejection in kidney transplant patients. Ultrasound-guided allograft biopsy is the best way to assess the reason for renal dysfunction. Because adverse reactions have been ascribed more frequently to the intravenous formulation, this route of administration is discontinued as soon as the patient can take the drug orally.
In rheumatoid arthritis, cyclosporine is used in severe cases that have not responded to methotrexate. Cyclosporine can be combined with methotrexate, but the levels of both drugs must be monitored closely. In psoriasis, cyclosporine is indicated for treatment of adult immunocompetent patients with severe and disabling disease for whom other systemic therapies have failed. Because of its mechanism of action, there is a theoretical basis for the use of cyclosporine in a variety of other T cell–mediated diseases. Cyclosporine reportedly is effective in Behçet’s acute ocular syndrome, endogenous uveitis, atopic dermatitis, inflammatory bowel disease, and nephrotic syndrome, even when standard therapies have failed.
Toxicity. The principal adverse reactions to cyclosporine therapy are renal dysfunction and hypertension; tremor, hirsutism, hyperlipidemia, and gum hyperplasia also are frequently encountered. Hypertension occurs in ~50% of renal transplant and almost all cardiac transplant patients. Hyperuricemia may lead to worsening of gout, increased P-glycoprotein activity, and hypercholesterolemia. Nephrotoxicity occurs in the majority of patients and is the major reason for cessation or modification of therapy. Combined use of calcineurin inhibitors and glucocorticoids is particularly diabetogenic. Especially at risk are obese patients, African American or Hispanic transplant recipients, or those with a family history of type II diabetes or obesity. Cyclosporine, as opposed to tacrolimus, is more likely to produce elevations in LDL cholesterol.
Drug Interactions. Any drug that affects CYPs, especially CYP3A, may impact cyclosporine blood concentrations. Substances that inhibit this enzyme can decrease cyclosporine metabolism and increase blood concentrations. These include Ca2+ channel blockers (e.g., verapamil, nicardipine), antifungal agents (e.g., fluconazole, ketoconazole), antibiotics (e.g., erythromycin), glucocorticoids (e.g., methylprednisolone), HIV-protease inhibitors (e.g., indinavir), and other drugs (e.g., allopurinol, metoclopramide). Grapefruit juice inhibits CYP3A and the P-glycoprotein multidrug efflux pump and thereby can increase cyclosporine blood concentrations. In contrast, drugs that induce CYP3A activity can increase cyclosporine metabolism and decrease blood concentrations. Such drugs include antibiotics (e.g., nafcillin, rifampin), anticonvulsants (e.g., phenobarbital, phenytoin), and others (e.g., octreotide, ticlopidine).
Interactions between cyclosporine and sirolimus require that administration of the 2 drugs be separated by time. Sirolimus aggravates cyclosporine-induced renal dysfunction, while cyclosporine increases sirolimus-induced hyperlipidemia and myelosuppression. Additive nephrotoxicity may occur when cyclosporine is coadministered with NSAIDs and other drugs that cause renal dysfunction; elevation of methotrexate levels when the 2 drugs are coadministered; and reduced clearance of other drugs, including prednisolone, digoxin, and statins.
ANTIPROLIFERATIVE AND ANTIMETABOLIC DRUGS
SIROLIMUS
Sirolimus (rapamycin; RAPAMUNE) is a macrocyclic lactone produced by Streptomyces hygroscopicus. Sirolimus inhibits T-lymphocyte activation and proliferation downstream of the IL-2 and other T-cell growth factor receptors (see Figure 35–1). Like cyclosporine and tacrolimus, therapeutic action of sirolimus requires formation of a complex with an immunophilin, in this case FKBP-12. The sirolimus–FKBP-12 complex does not affect calcineurin activity; rather, it binds to and inhibits the protein kinase mTOR
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


