| USAN: | Imatinib Mesylate |
| Brand Name: | Gleevec (Novartis) |
| Molecular Weight: | 589.71 (Parent, 493.60) |
| FDA Approval: | 2001 |
| Drug Class: | Protein-tyrosine Kinase Inhibitor |
| Indications: | Treatment of Chronic Myeloid Leukemia (CML) and Gastrointestinal Stromal Tumors (GIST) |
| Mechanism of Action: | Inhibiting a Set of More Than 8 Protein-Tyrosine Kinases |
Imatinib mesylate (1, Gleevec) is a protein-tyrosine kinase inhibitor for the treatment of chronic myeloid leukemia (CML) and gastrointestinal stromal tumors (GIST). It is the first orally bioavailable anticancer drug that only targets enzymes (protein kinases) that are specific to tumor cell growth but not required by healthy cells. Therefore, protein kinase inhibitors are also known as targeted cancer drugs or smart drugs.
Gleevec was Novartis’s top performer in 2012 with sales of $4.6 billion.
■ 1 History
1.1 A Brief History of Cancer Drugs
Great strides had been made in the war against cancer with chemotherapy even before the emergence of protein kinase inhibitors.1 For instance, prior to vinblastine (1, Velban) became available in 1964 for the treatment of lymphoma, the diagnosis of Hodgkin’s disease (a cancer of the lymph nodes) was virtually a death sentence. Today there is a 90% chance of survival with the treatment by vinca alkaloids such as 1 and other chemotherapies. Similarly, when Sidney Farber discovered the effects of methotrexate (2, Trexall) on leukemia, it marked the beginning of the triumph over childhood leukemia.

Following Barnett Rosenberg’s discovery of cisplatin (3, Platinol)’s effects on tumor cells in 1967, cisplatin and its analogs such as carboplatin (4, Paraplatin) and oxaliplatin (5, Eloxatin) contributed significantly in boosting the survival rate of patients with metastatic testicular cancer, ovarian tumors, and bladder cancer. Most significantly, breast cancer, a malady striking one in eight women, has been effectively managed via a plethora of treatments including surgery, radiation, and chemotherapies. The arsenal of chemotherapeutics for treating breast cancer includes SERMs such as tamoxifen (6) and raloxifene (7, Evista). Type I, II, and III aromatase inhibitors have now also been widely prescribed to combat breast cancers (more details may be found in chap. 4). Today, breast cancer is sometimes viewed as a chronic disease that can be managed, rather than a lethal disease.


Despite the efficacy of the aforementioned chemotherapeutics, they kill cancer cells and normal cells with equal ferocity. (Some have compared chemotherapy to a “carpet bombing” strategy.) However, the reason these chemotherapies are effective is that cancer cells divide at much faster rate than normal cells; therefore, chemotherapies kill more malignant cells than healthy cells.
Chemotherapies invariably come with significant side effects rooted. For example, hair follicle cells have a physiologically high mitosis rate; therefore, chemotherapies kill them faster than other healthy cells. In the same vein, other common side effects of chemotherapy include diarrhea (because ephithelial renewal is inhibited), bone marrow suppression (because granulopoiesis, thrombopoiesis, cytopoiesis, and erythropoiesis are inhibited), and lymph node damage (because of lymphocyte multiplication inhibition causes immune weakness). These toxicities manifest as damage to the gastrointestinal tract, bone marrow, nervous system, kidneys, heart, or pancreas. Therefore, a cancer treatment that kills the cancer cells while leaving the healthy cells alone is highly desirable. In other words, a strategy with the same or better efficacy as chemotherapy but without its toxicities is needed.
Protein kinase inhibitors fit the bill. In contrast to the “carpet bombing” strategy of chemotherapy, protein kinase inhibitors are akin to “precision bombs” that only kill cancer cells, and are thus devoid many of the toxicities (and therefore the side effects) that plagued conventional chemotherapies.
1.2 Treatments of Chronic Myeloid Leukemia
Among the many subtypes of leukemia, a rare but particularly ferocious form is chronic myeloid leukemia (CML) where “myeloid” indicates that it is marrow-related. CML is a hematologic stem cell disorder caused by an acquired abnormality in the DNA of the stem cells in bone marrow and is characterized by abnormally high white blood cell counts.2 CML is one of the four main types of leukemia, striking about 5,000,000 patients a year in the US. It mainly affects adults between the ages of 50 and 60.
CML was first described by Rudolf Virchow, among others, around 1845. In 1872, Ernst Neumann observed that CML cells originated in the bone marrow. One of the early treatment options for treating CML was a bone marrow transplant. However, not only was a matching donor needed, but the procedure was highly risky with a survival rate of only 50%. Effective control of blood counts became feasible in 1959 with the orally bioavailable alkylating agent busulfan (8, butane-1,4-bis-mesylate) synthesized by Geoffrey Timmis at Burrroughs Wellcome. Busulfan acts on primitive stem cells and is rarely used today because of its severe, toxic side effects. The better-tolerated hydroxyurea (9), developed by BMS and the University of Chicago, became available in 1969. A ribonucleotide reductase inhibitor, hydroxyurea (9) became a commonly used cytotoxic agent in the treatment of CML thanks to both the fast onset of its effect and relatively low toxicities. Soon afterward, interferon-α was found to induce durable major cytogenetic responses (decreasing the number of Philadelphia chromosome cells) and long-term survival, although in only a small fraction of patients, reducing the white cell count to tolerable levels. It became for a while the most popular treatment for CML. Unfortunately, in addition to being a biologic that has to be given intravenously, the flu-like side effect brought on by interferon makes it less desirable. Another intravenous drug, cysteine arabinose (10, Ara-C, cytarabine), came along later. Finally, cortisone (11) and prednisone (12) at high doses are also known to kill leukemic lymphocytes and lymphoblasts.


Again, drugs 8–12 and interferon are all chemotherapy, and all possess significant toxicities. Conversely, Novartis’s Gleevec (1) specifically targets a protein kinase Abl (short for “Abelson; see below) for the treatment of CML. Gleevec (1) is the first marketed drug whose MOA is via inhibition of a protein kinase, functioning as a signal transduction inhibitor (STI). Imatinib represents a new paradigm in cancer therapy, revolutionizing the treatment of CML and GIST patients as an oral drug with side effects that are both relatively fewer and more tolerable than those of hydroxyurea (9), Ara-C (10), and interferon.
In 1960, Philadelphia cytogeneticists Peter Nowel and David Hungerford described an abnormally small G-group chromosome that we now call the Philadelphia chromosome (Ph) in CML patients. This was a seminal discovery as it unequivocally proved that CML cancer was directly linked to an abnormality of DNA.
In 1970, Herbert Abelson, working with David Baltimore, isolated a transforming gene from a specific variant of the Moloney leukemia virus. In 1973, Janet Rowley recognized that Ph was the product of a reciprocal translocation between chromosomes 9 and 22. In the 1980s, the human Abelson gene (Abl) was found on human chromosome 9 but translocated to the Ph chromosome in CML. Also in the 1980s, the translocation partners were identified as breakpoint cluster region (Bcr) and Abl, followed by the discovery that unregulated tyrosine kinase activity is critical to Bcr–Abl’s ability to transform cells. The disappearance of the Ph chromosome in CML patients who underwent allotransplants, the first cures of CML, was also reported.
In 1986 and 1987, David Baltimore and his postdoctoral fellow, Owen Witte, published two articles in Science identifying Bcr–Abl as a tyrosine kinase. Like epidermal growth factor receptor (EGFR), Bcr–Abl is an enzyme that carries out signal transduction through the transfer of phosphate groups to specific amino acids (tyrosine in this case) on a protein. As a consequence, the cells receiving the signal begin dividing uncontrollably, thus triggering cell proliferation and leukemia. The hallmark of CML is the expression of Bcr–Abl. In effect, Baltimore and Witte identified the Bcr–Abl gene as the cause of CML. It then made sense that blocking the Bcr–Abl enzyme and stopping the faulty signal transduction would stave off the production of white blood cells. CML is one of very few cancers that has been directly linked to a single oncogene (i.e., Bcr–Abl). Most cancers are associated with at least two, and often more, oncogenes. In 1990, a faithful murine disease model for CML was established, which greatly boosted the research in the field. In 1992, Alexander Levitzki from Israel proposed the use of Abl inhibitors to treat leukemia driven by Abl oncogenes. In reality, at that time many hurdles still existed on the road to an oral drug for combating CML.
1.3 Protein Kinase Inhibitors
In 1966 Fischer and Krebs published their seminal work on protein phosphorylation (i.e., adding a phosphate) and its regulatory function in cellular pathways.3 As shown in Fig. 5.1, protein kinases modulate intracellular signal transduction by catalyzing the phosphorylation of specific proteins. For example, protein phosphorylation catalyzed by protein kinase C (PKC) is activated by diacylglycerol (DAG). On the contrary, protein phosphatases function through de-phosphorylation of proteins to modulate biological activities, while concomitantly ATP (adenosine triphosphate, 13) loses one molecule of phosphate to give ADP (adenosine diphosphate, 14). Many cellular processes are the result of the interplay between phosphorylation by protein kinases and dephosphorylation by protein phosphatases.
Protein kinases comprise more than 518 members of ATP-regulated cellular or membrane-bound proteins that are capable of donating phosphate groups to target proteins.4 They regulate critical cellular processes including gene transcription and cell growth, proliferation, and differentiation, often through complex and interactive processes. Since protein kinases are responsible for signal transduction, turning on and off the switches for cancer cells to grow, they are strongly implicated in cancer. Oncogenic kinases5,6 are known to cause aberrant and often uncontrollable cellular phosphorylation and lead to tumor formation. By blocking the functions of protein kinases, it may be possible to stop cancer growth. Therefore kinase inhibitors hold great potential as anticancer therapeutics.


Fig. 5.1. The Functions of Protein Kinase
Protein kinases can be classified according to the amino acid residue that is phosphorylated in the cellular process. Consequently, there are tyrosine-specific kinases and serine/threonine kinases. Tyrosine kinases are a family of tightly regulated enzymes involved in phosphorylation to tyrosine, and the aberrant activation of various members of this family is one of the hallmarks of cancer. Tyrosine phosphorylation has been linked to multiple cell growth and differentiation pathways. Therefore, protein kinase inhibition is an area for therapeutic intervention against a variety of diseases such as cancer, inflammatory disorders, and diabetes.
Because protein kinases are selective against cancer cells, it is hoped that protein kinase inhibitors will have good efficacy without the toxicities associated with older chemotherapies. They promise a kinder, gentler, and more effective method for cancer treatment. Hence, protein kinase inhibitors are called targeted cancer drugs.
As a testimony to the importance of the protein kinase field, Bishop and Varmus received the 1989 Nobel Prize (for Physiology or Medicine) for their work on oncogenic kinases.7 In 2001, Nurse and Hunt, along with Hartwell, were also awarded the Nobel Prize for their work in elucidating the role of cyclins and cyclin-dependent kinases (CDK) in regulating cell cycles.8
Biologics as protein kinase inhibitors were the first approved cancer treatments by inhibiting kinases. The first three are trastuzumab (Herceptin), cetuximab (Erbitux), and bevacizumab (Avastin). As of 2014, more than 12 monoclonal antibodies (mAbs) have been approved by the FDA for the treatments of cancers.
Genentech’s Herceptin is a bioengineered humanized monoclonal antibody (shown in Fig. 5.2) for treating breast cancer. It was the first targeted cancer drug. Herceptin inhibits a human cellular oncogene—epidermal growth-factor receptor-2 (EGFR-2), also known as the HER-2/neu gene. Trastuzumab (Herceptin) was 95%t human and 5% murine (it is known as a chimeric antibody). It was approved by the FDA for treatment of HER-2 over-expressed metastatic breast cancers in 1998.9

Fig. 5.2. From Murine to Humanized mAb
Cetuximab (Erbitux) is a chimeric monoclonal antibody of EGF receptors, which are found in a third of solid cancer tumors. It was approved by the FDA for clinical use in 2004 to treat EGFR-expressing metastatic colorectal cancer.10 Genentech’s Avastin vindicated the anti-angiogenesis approach to fighting cancer. Avastin, a humanized monoclonal antibody against vascular endothelial cell growth factor (VEGF) receptor,11 is the first drug to be marketed that works through the anti-angiogenesis mechanism.12 It is 96% human and 4% murine.13
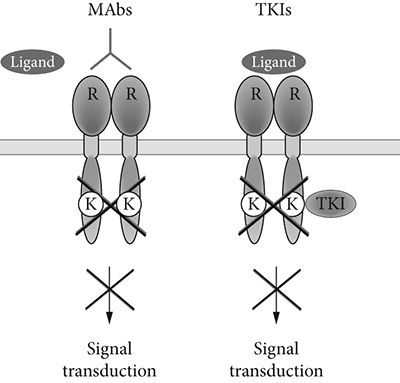
Fig. 5.3. Monoclonal antibodies (MAbs) vs. Tyrosine Kinase Inhibitors (TKIs)
The success of the aforementioned monoclonal antibodies in fighting cancers opened the floodgates to using small-molecule protein kinase inhibitors as oral drugs. Monoclonal antibodies left much to be desired since they are very expensive to make—manufactured by growing an enormous number of genetically identical ovary cells from hamsters in huge vats. In addition, because they are large proteins that do not survive in the gut they can only be administered by infusion. Oral drugs would, on the other hand, be cheaper to make and easier to swallow, so to speak.
So far, clinical studies have begun on hundreds of small-molecule kinase inhibitors. Most of these target the highly conserved ATP-binding site, leading to concerns about kinase selectivity. However, more than 20 small-molecule kinase inhibitors have overcome this early skepticism and have achieved regulatory approval as targeted antitumor therapeutics. As shown in Fig. 5.3, monoclonal antibodies bind to the receptors at the exterior of the cell’s membrane whereas small molecule protein kinase inhibitors penetrate the cell’s membrane and block the enzyme’s function in the interior of the cell. Interestingly, most kinase inhibitors are flat aromatic compounds that mimic the adenine portion of ATP (13).
The first (in 2001) small-molecule kinase inhibitor on the market was Gleevec (1) for the treatment of CML and GIST. The second was AstraZeneca’s gefitinib (Iressa, 15). An EGFR inhibitor, Iressa was launched in 2003 for the treatment of locally advanced or metastatic non–small-cell lung cancer. While Iressa’s efficacy for Caucasian cancer patients saw mixed results, a subset analysis from a large clinical trial showed robust efficacy in patients of Asian origin with refractory advanced non–small-cell lung cancer.14

Sunitinib maleate (16, Sutent, SU11248) was discovered by Sugen, which later became part of Pfizer.15 Sutent is an orally active drug that exhibits potent anti-angiogenic activity through the inhibition of multiple receptor tyrosine kinases (RTKs). Specifically, 16 inhibits vascular EGFR, vascular endothelial growth factor receptor1 (VEGFR1), VEGFR2, and VEGFR3 and platelet-derived growth factor receptors-α (PDGFR-α) and PDGFR-β. In addition, Sutent also targets receptors implicated in tumerogenesis including fetal liver tyrosine kinase receptor 3 (Flt3) and stem-cell factor receptor (c-KIT). A drug with a multitarget profile is less likely to lead to drug resistance than a compound that displays selectivity for a single kinase. Furthermore, drugs that act on multiple pathways simultaneously may be more efficacious than single-target agents.

Unfortunately, although the response rates for patients on first-line therapy with imatinib (1) are very high, approximately 22–41% of patients develop resistance to it—a significant percentage due to specific mutation in the Bcr–Abl kinase domain. Dasanitib (17, Sprycel), a dual Bcr–Abl and Src kinase inhibitor, is a good choice as a second-line tyrosine kinase inhibitor in Gleevec-resistant CML patients harboring Bcr–Abl kinase domain mutations.16 It is likely that dasanitib (17) binds to different loops from those of Gleevec (1).

Nilotinib (18, Tasigna), Novartis’s follow-up to Gleevec (1), is an attempt to treat Gleevec-resistant CML patients.17

In addition to 1 and 15–18, there are about a dozen additional small-molecule protein kinase inhibitors that have been approved by the FDA for the treatment of cancer. They include axitinib (Inlyta, Pfizer, 2012), bosutinib (Bosulib, Pfizer, 2012), cabozantinib (Cometriq, Exelixis, 2012), cetuximab (Erbitux, ImClone, 2006), crizotinib (Xalkori, Pfizer, 2011), erlotinib (Tarceva, Genentech, 2005), lapatinib (Tykerb, GSK, 2007), pazopanib (Votrient, GSK, 2009), ponatinib (Iclusig, Ariad, 2012),18 ruxolitinib (Jakafi, Incyte, 2011), sorafenib (Nexavar, Bayer, 2005),19 vemurafenib (Zelboraf, Roche, 2011). Among these, bosutinib (Bosulib) is a promising candidate for third-line use in patients who fail on second-line TKI (tyrosine kinase inhibitor) therapy, and is being explored as a second-line therapy in its own right.
1.4 Genesis of Gleevec
In 1985, Alex Matter of Ciba-Geigy in Basel, Switzerland embarked on an ambitious oncology program targeting kinases. He and his colleague Nick Lydon did not make much progress until Brian Druker at Dana-Farber suggested that Ciba-Geigy work on targeting the Bcr–Abl kinase for CML in 1988. At the time it was already known that Bcr–Abl is an abnormal protein tyrosine kinase produced by the specific chromosomal abnormality called the Philadelphia chromosome and that this chromosome is a marker for CML. Two additional pieces of information aided their work: A few years earlier, Japanese scientists showed that staurosporine (19), an indole natural product, inhibited the receptor of PKC.20 And, in 1988, Alexander Levitzki showed that selectivity could be achieved for EGFR. In a Science paper,21 Levitzki revealed that a series of simple dicyanobenzylidenes such as 20 and carboxybenzylidenes could block EGF-dependent cell proliferation. The dicyanobenzylidenes were among the first selective small molecule EGFR kinase inhibitors.

There are more than 500 protein kinases, which all bear an uncanny structural resemblance to each other. To create a molecule that selectively targets Bcr–Abl required not only ingenuity, but also fantastic luck. An unselective inhibitor would have undesirable toxicity. In addition, since most protein kinase inhibitors resemble the adenosine molecule on ATP, they are mostly flat molecules and are notoriously insoluble; insoluble compounds cannot penetrate the cell membranes. The Bcr–Abl protein is located on the interior of cells, whereas the EGFR protein is extracellular, extruding out of the surface of cell membrane; thus it is more accessible. To bind to the Bcr–Abl protein, the drug has to penetrate the cell membrane or it will be completely useless.
Phenylaminopyrimidine 21 was initially identified by Ciba-Geigy from a high through-put screening (HTS) as a PKC-α inhibitor with an IC50 of approximately 1 μM. It did not inhibit Abl and PDGFR. Intensive SAR studies involving more than 300 analogs eventually led to STI571, which later became Gleevec (1) (Fig. 5.4). After STI571 was made, protein binding and later cellular assays by the biologists revealed that not only was it a potent blocker for the phosphorylation of the Bcr–Abl kinase, it was also selective against most of the major kinases that they screened. In 1993, Druker also tested STI571 in protein and cellular models. He observed that STI571 killed CML cells without harming the healthy cells. STI571 subsequently showed sufficient efficacy in animal (murine) models.

Fig. 5.4. Evolution of the screening lead 21 to imatinib (1)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


