| USAN: | Amlodipine Besylate |
| Brand Name: | Norvasc (Pfizer) |
| Molecular Weight: | 567.06 (Parent 408.88) |
| FDA Approval: | 1990 |
| Drug Class: | 1,4-Dihydropyridine Calcium Channel Blocker |
| Indications: | Treatment of Hypertension and Angina Pectoris |
| Mechanism of Action: | Calcium Channel Blocker (Inhibition of the Trans-membrane Influx of Calcium Ions into Vascular Smooth Muscle and Cardiac Muscle with Peripheral Arterial Vasodilatation) |
■ 1 History
Hypertension (high blood pressure) is estimated to afflict 1 billion individuals worldwide and is a major risk factor for stroke, coronary artery disease, heart failure, and end-stage renal disease.1 The first class of drugs to treat hypertension was the mercurial diuretics, discovered by Alfred Vogl in 1919 in Vienna.2 Diuretics, by removing fluid from the body, reduce the pressure on the heart. Mercurial diuretics revolutionized the treatment of congestive heart failure resulting from severe edema and were the primary treatment until the late 1950s, when thiazide diuretics emerged. In 1957, Merck chemist Frederick C. Novello prepared chlorothiazide (2, Diuril), a potent diuretic that does not cause elevation of bicarbonate excretion, an undesired side effect associated with mercurial diuretics.3 Shortly after chlorothiazide’s (2) success, George deStevens at Ciba reduced a double bond on chlorothiazide (2) to a single bond to give hydrochlorothiazide (3, HydroDiuril) which was 10-fold more potent than the prototype 2.4 Hydrochlorothiazide was introduced to medical practice in 1959 and within a short time became the drug of choice for the treatment of mild hypertension.
One of the liabilities of these drugs is thiazide diuretic-induced hyperglycemia.

As early as 1948, Raymond P. Ahlquist at the Medical College of Georgia speculated that there were two types of adrenergic receptors (adrenoceptors in short), which he termed α-adrenoceptor and β-adrenoceptor, that are 7-transmembraned protein as GPCR.5 In 1957, Irwin H. Slater and C. E. Powell at Eli Lilly prepared dichloroisoprenaline (DCI, 4), the dichloro analog of isoprenaline; it was later found to be the first selective β-adrenoreceptor blocking reagent, also known as a β-blocker. However, DCI was not further pursued as a drug candidate because it had a marked undesirable stimulant effect on the heart, an intrinsic sympathomimetic action (ISA).6 Beginning in 1958, James Black at Imperial Chemical Industries (ICI) led a team to look for β-blockers that were devoid of the stimulant effect on the heart. In 1962, the first selective β-adrenoreceptor inhibitor pronethalol (5) was discovered but was withdrawn from further development when it was found to cause thymic tumors in mice. ICI eventually in 1964 produced a drug propranolol (6, Inderal), which possessed a better efficacy and safety profile.7 Propranolol (6) is now widely used in the management of angina, hypertension, arrhythmia, and migraine headaches. Two additional β-blockers, atenolol (Tenormin) and practolol (Dalzic), were later discovered and marketed by ICI. Dozens of “me-too” selective beta-blockers have since been discovered and marketed.

Angiotensin converting enzyme (ACE) inhibitors are widely used to treat hypertension, congestive heart failure, and heart attacks. The MOA of ACE inhibitors is through inhibiting ACE in the renin–angiotensin system (RAS), which is the master regulator of blood pressure and cardiovascular function. RAS provided numerous targets for pharmacologic intervention (Fig. 3.1).11 Although renin catalyzes the first and rate-limiting step (see 12 below) in the activation of the RAS, it was the inhibition of the downstream ACE that first established the clinical relevance of this pathway in the treatment of hypertension.
David Cushman and Miguel A. Ondetti at the Squibb Institute isolated teprotide, a nonapeptide, from the poisonous venom extract of the Brazilian pit viper Bothrops jararaca. Using teprotide, a potent ACE inhibitor in vitro, as a starting point, Cushman and Ondetti curtailed the molecule, replaced its carboxylate group with a thiol (–SH), and achieved a 2,000-fold increase in potency in ACE inhibition. The resulting drug became the first oral ACE inhibitor, captopril (7, Capoten).8 Merck scientists led by Arthur A. Patchett discovered the second oral ACE inhibitor, enalapril (8, Vasotec), which has been on the market since 1981.9 Another popular ACE inhibitor is Parke-Davis’s quinapril hydrochloride (9, Accupril), launched in 1991.10


Also as shown in Fig. 3.1, angiotensin II is a potent vasoconstrictor; blocking its action would result in vasodilation. DuPont-Merck Pharmaceuticals discovered the first inhibitor of the angiotensin II receptor, losartan (10, Cozaar), which after its launch in 1995 quickly became one of the most important drugs for the treatment of high blood pressure.12 Other angiotensin II receptor antagonists (also known as angiotensin receptor blockers, or ARBs) include Novartis’s valsartan (11, Diovan),12 Sanofi-Aventis’s irbesartan (Avapro), AstraZeneca’s candesartan (Atacand), and Boehringer-Ingelheim’s telmisartan (Micardis). They all proved to be superior to ACE inhibitors because they did not cause the irritating cough that occurs in a small percentage of patients taking ACE inhibitors.

Since renin is extremely specific for angiotensinogen and is the first and rate-limiting enzyme of the RAS, renin inhibition was recognized for decades as an attractive approach for the treatment of hypertension and hypertension-related target organ damage. Novartis’s aliskiren (12, Tekturna) is the first and the only renin inhibitor on the market for treatment of hypertension, and has been since 2007.11


The concept of calcium channel blockers (CCBs), also known as calcium channel antagonists or calcium entry blockers, was developed several years after some CCBs including phenylalkylamines such as verapamil (13), perhexiline, and prenylamine; and benzodiazepines such as diltiazem (14) were discovered. Albrecht Fleckenstein at University of Freiburg in Germany and Winifred G. Nayler helped to elucidate the mechanism of action (MOA) of those structurally diversified drugs as CCBs.13
In 1969, Bayer Company investigated the pharmacology of Bay-a-1040 (15, nifedipine) and Bay-a-7168 (niludipine). Both compounds were strong coronary vasodilators and exerted significant negative inotropic effects on the myocardium. With Fleckenstein’s help, Bayer elucidated their MOA as CCBs. Nifedipine (15, Adalat) heralded the beginning of one of the most important classes of calcium antagonists: 1,4-dihydropyridines.13

Bayer’s nifedipine (15, Adalat), known as a first-generation CCB, is a short-acting CCB with a short plasma half-life (in the range of 0.5 to 2 h). Therefore, Adalat has to be taken 3 or 4 times a day to elicit 24-h blood pressure control. Efforts to improve bioavailability resulted in the second-generation CCBs, including isradipine (t1/2 ~2 h), nicardipine (t1/2, ~5 h), and felodipine (t1/2, ~10 h). Pfizer’s amlodipine (1, Norvasc), launched in 1990, belongs to the third-generation of CCBs. It has a high bioavailability (64%) and a longer half-life (~45 h) in plasma, so it can be taken once daily.14 All of these features made amlodipine (1, Norvasc) the most prescribed antihypertensive agent in the world in 2003 with an annual sale of $4.3 billion. The worldwide sales of calcium channel blockers totaled $6 billion that year.
■ 2 Pharmacology
2.1 Mechanism of Action
Inside a normal cell, the concentration of free Ca2+ ions is low (10–7 M) in comparison to that of the extracellular fluid (1–2 mM). The concentration may be regulated by opening or closing of the calcium channels. Calcium channels are opened temporarily by exogenous impulses, whereby the calcium ion concentration rises briefly and Ca2+ ions are bound to calcium-binding proteins (calmodulin). The activated calmodulin elicits actual reactions in the cell and the increased calcium concentration is reduced rapidly to the initial value by uptake of Ca2+ ions into intracellular reserves.
The calcium channel, located on the cell membrane, is an ion channel that is selectively permeable to calcium ions (Fig. 3.2). Since the results of calcium ions entering the cell membrane include contraction of smooth muscle cells, blocking the entrance of calcium ions into the cell would result in vasodilation, thus lowering the blood pressure. When a CCB enters the opening of a calcium channel, the drug figuratively gets stuck, like a fat man caught halfway through a porthole, thus preventing calcium ions from getting through the channel.
Amlodipine (1, Norvasc) and other CCBs inhibit the influx of Ca2+ ions into cells without affecting inward Na+ or outward K+ currents to a significant degree. The sizes of Ca2+, Na+, and K+ are quite different so selectivity might be relatively easier to achieve if the drug is not too small.
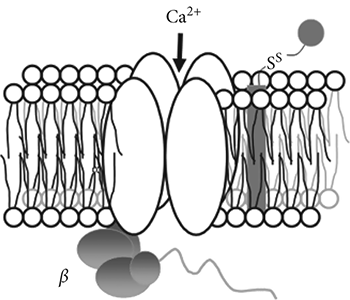
Fig. 3.2. The Calcium Channel
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


