Fig. 6.1
Immunofluorescence microscopy demonstrating glomerular mesangial staining for immunoglobulin A (IgA) in a patient with IgA nephropathy
Pathologic Findings
Light Microscopy
IgA nephropathy and IgA vasculitis (Henoch-Schönlein purpura) nephritis can have any of the histologic phenotypes of immune complex-mediated glomerulonephritis other than pure membranous glomerulopathy, including no lesion by light microscopy with immune deposits by immunohistology, mesangioproliferative glomerulonephritis with mesangial but no endocapillary hypercellularity (Figs. 6.2 and 6.3), focal or diffuse proliferative glomerulonephritis with endocapillary hypercellularity (with or without crescents) (Fig. 6.4), overt crescentic glomerulonephritis with 50 % or more crescents, type I membranoproliferative (mesangiocapillary) glomerulonephritis (rare), and focal or diffuse sclerosing glomerulonephritis [4, 7–10].
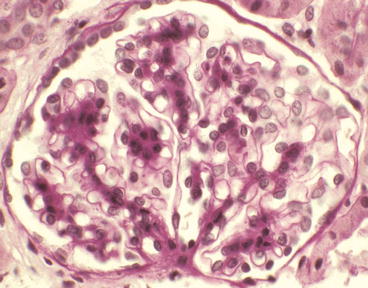
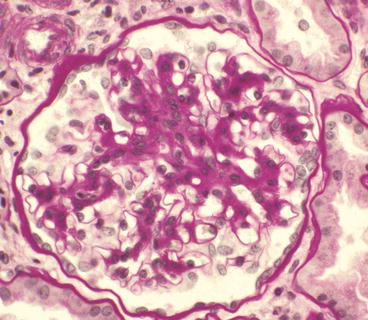
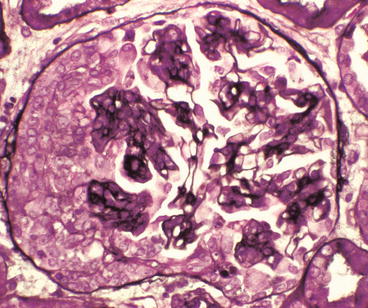
Fig. 6.2
Glomerulus from a patient with IgA nephropathy showing mild segmental mesangial hypercellularity in the upper left quadrant of the glomerulus [periodic acid-Schiff (PAS) stain]
Fig. 6.3
Glomerulus from a patient with IgA nephropathy showing moderate segmental mesangial hypercellularity and increased mesangial matrix in the upper portion of the tuft (PAS stain)
Fig. 6.4
Glomerulus from a patient with Henoch-Schönlein purpura showing a proliferative glomerulonephritis with endocapillary hypercellularity adjacent to a cellular crescent on the left of the tuft (Jones silver stain)
A variety of classification systems have been used to categorize the light microscopic phenotypes of IgA nephropathy, such as those proposed by Kurt Lee et al. [11] and by Mark Haas [12] (Table 6.1). Another approach is to use the same descriptive terms (but not the numerical class designations) that are in the lupus classification system to categorize IgA nephropathy as well as other forms of immune complex glomerulonephritis. This terminology works as well for IgA nephropathy as it does for lupus and also has the advantage of not requiring knowledge of multiple different classification systems. Recently, a novel classification approach (the Oxford Classification System) has been developed [4]. This will be discussed later in this chapter.
Table 6.1
Three different approaches to the histologic classification of IgA nephropathy
Lee system | Haas system | Lupus terminology |
|---|---|---|
I: Focal mesangioproliferative | I: Focal mesangioproliferative | No lesion by light microscopy (I) |
II: Moderate focal proliferative | II. Focal proliferative | Mesangioproliferative (II) |
III: Mild diffuse proliferative | III: Focal sclerosing | Focal proliferative (III) |
IV: Moderate diffuse proliferative | IV: Diffuse proliferative | Focal sclerosing (III C) |
V: Severe diffuse proliferative | V: Chronic sclerosing | Diffuse proliferative (IV) |
Chronic sclerosing (VI) |
In patients whose renal biopsy specimens are referred to the University of North Carolina for evaluation, crescents are observed in about a third of patients with IgA nephropathy and two thirds of patients with IgA vasculitis nephritis (Henoch-Schönlein purpura) [13]. However, overt crescentic glomerulonephritis with 50 % more of glomeruli with crescents is uncommon (<5 % in IgA nephropathy and <10 % in IgA vasculitis nephritis). When substantial crescent formation is present, especially with conspicuous fibrinoid necrosis, the possibility of concurrent antineutrophil cytoplasmic antibody (ANCA) disease should be considered [14].
Between 5 and 10 % of specimens with IgA nephropathy identified by immunohistology have focal segmental glomerulosclerosis as seen on light microscopy that is indistinguishable from idiopathic focal segmental glomerulosclerosis [12]. Most instances of this pattern of injury probably are glomerular scarring caused by earlier active focal segmental glomerular inflammation rather than another form of secondary focal segmental glomerulosclerosis (FSGS). However, at least some focal segmental glomerular sclerosis in IgA nephropathy may be caused by compensatory hemodynamic changes following loss of nephrons or through podocyte damage secondary to mediators released from activated mesangial cells [15, 16].
Immunofluorescence Microscopy
The sine qua non for a diagnosis of IgA nephropathy is immunohistologic detection of dominant or codominant staining for IgA in the glomerular mesangium (Fig. 6.1). A caveat to this is that the staining for IgA should at least be 1+ on a scale of 0–4+ or 0–3+. Trace amounts of IgA are not definitive evidence for IgA nephropathy [4]. The IgA is predominantly IgA1 rather than IgA2 [7, 17]. Capillary wall staining is observed in about a third of patients and is more common in IgA vasculitis (Henoch-Schönlein purpura) nephritis [10]. The mesangial immune deposits of IgA nephropathy stop abruptly at the glomerular hilum and are not observed along tubular basement membranes. Rare patients have IgA nephropathy concurrent with membranous glomerulopathy, and thus, their specimens show granular capillary wall IgG staining and mesangial IgA-dominant staining [18, 19].
Staining for IgA frequently is accompanied by staining for other immunoglobulins and complement components [3]. Staining for IgG and IgM often is present but at lower intensity compared to IgA. A very distinctive feature of IgA nephropathy compared to other immune complex diseases is the predominance of staining for lambda over kappa light chains in many specimens. C3 staining is almost always present and usually relatively bright. However, staining for C1q is uncommon and when present is typically of low intensity. The presence of substantial C1q should raise the possibility of lupus nephritis with conspicuous IgA deposition. This suspicion would be supported further by finding endothelial tubuloreticular inclusions by electron microscopy and antinuclear antibodies serologically. As in other forms of glomerulonephritis, staining for fibrin is seen at sites of necrosis and crescent formation. Depending in part on what reagent antibody is used, the immune deposits occasionally stain for fibrin, especially in patients with IgA vasculitis (Henoch-Schönlein purpura) nephritis [10]. The IgA deposits stain predominantly for IgA1 rather than IgA2 and also stain for secretory component in about a third of specimens [17].
Glomerular diseases other than IgA nephropathy that can have IgA-dominant or IgA-codominant deposits include lupus glomerulonephritis, rare examples of IgA-dominant anti-GBM disease [20, 21], and an IgA-dominant form of postinfectious glomerulonephritis that is usually caused by staphylococcal infections [5].
Electron Microscopy
The typical ultrastructural finding is immune complex-type electron-dense deposits in the mesangium (Figs. 6.5 and 6.6). Dense deposits most often are found immediately beneath the paramesangial glomerular basement membrane. The amount of deposits varies substantially, with occasional specimens having massive replacement of the matrix by the dense material (Fig. 6.6). Rare specimens that have well-defined IgA deposits by immunofluorescence microscopy do not have detectable mesangial dense deposits, which does not rule out a diagnosis of IgA nephropathy because the immunohistology is the defining feature. Capillary wall subepithelial, subendothelial, and intramembranous deposits are identified in approximately a quarter to a third of specimens with IgA nephropathy [3] and are more frequent in patients with IgA vasculitis (Henoch-Schönlein purpura) nephritis [10]. Capillary wall deposits are least frequent in histologically mild disease and most frequent in histologically severe disease, especially when crescents are present.
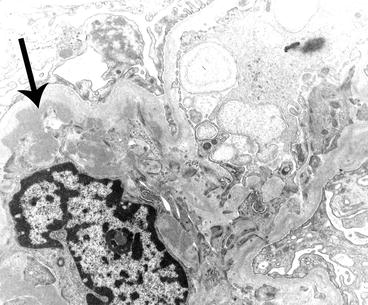
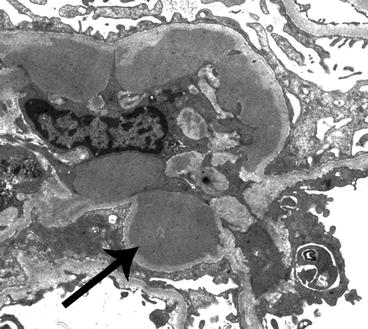
Fig. 6.5
Electron micrograph of a glomerulus from a patient with IgA nephropathy showing a moderate amount of electron-dense deposits (arrow) within the mesangium. The mesangium is on the left of the image and a portion of the capillary loop is on the right
Fig. 6.6
Electron micrograph of a glomerulus from a patient with IgA nephropathy showing massive electron-dense deposits (arrow) within the mesangium. The mesangium is on the left of the image and a portion of the capillary loop is on the right
Focal areas of glomerular basement membrane thinning are observed in approximately a third of specimens with IgA nephropathy [22]. This structural abnormality may contribute to the hematuria. Focal or diffuse podocyte foot process effacement often is present, especially in patients with nephrotic range proteinuria. Foot process effacement is particularly prominent in patients who have the syndrome of histologically mild IgA nephropathy with minimal change glomerulopathy-like features clinically [23]. Mesangial matrix expansion and mesangial hypercellularity parallel the mesangial changes seen by light microscopy.
Etiology/Pathogenesis
Multiple pathogenic factors and multi-hit combinations of factors probably contribute to the development of IgA nephropathy and IgA vasculitis [6, 7, 24, 25]. A frequent if not ubiquitous pathogenic factor is abnormal structure and function of IgA molecules resulting from aberrant glycosylation of IgA1 hinge regions [7]. This abnormality could result in mesangial IgA deposition by a variety of mechanisms including reduced clearance from the circulation because of lack of receptor engagement by the abnormal IgA, increased aggregation of IgA in the circulation resulting in mesangial trapping, development of immune complex-forming IgG autoantibodies directed against the abnormal IgA, increased affinity of the abnormal IgA for mesangial matrix, or combinations of these processes. Other factors could induce or act synergistically with this glycosylation abnormality. For example, a mucosal infection could induce excessive production of aberrant IgA1 in patients who are genetically determined to make abnormally glycosylated IgA1. Alternatively, mucosal infectious pathogens could release enzymes (e.g., neuraminidase, sialidase) that induce nephritogenic alterations in IgA1 glycosylation. These effects by infections could explain the close association between the onset and exacerbations of IgA nephropathy with respiratory and gastrointestinal tract infections.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


