Ideal gases
A frequently used approach in physical chemistry is to define an ideal system: a system that is an approximation to a real system, in which the properties can be simply explained. Real systems – those that are actually encountered in the lab or in Nature – are often complex and not fully understood. Our starting point is, therefore, to define a system we can describe exactly. We then determine how good our ideal system represents real systems, over what ranges the ideal system describes real systems accurately, and the limits of its applicability. We need to determine whether, even if it does not deliver quantitatively accurate results, it still describes trends accurately; and whether there are simple corrections that can be added on to enhance the quantitative agreement and range of applicability of the ideal system. Molecular interactions are complex. Therefore, we begin by considering the gas phase, in which molecules are far apart on average. This minimizes the importance of intermolecular interactions compared to condensed phases (liquids and solids). Indeed, our first assignment is to define an ideal gas in which there is a complete absence of intermolecular interactions. Here again we use the term molecule to refer to the particles in the gas, regardless of whether they are atoms or molecules.
An ideal gas has the following properties:
- The gas is composed of point-like molecules.
- At any finite temperature the molecules are in constant motion and collide elastically with each other and the walls of their container.
- There are no intermolecular interactions (neither attractions nor repulsions).
- An ideal gas follows the equation of state pV = nRT.
The first point means that even though a sample of gas, which contains n moles of molecules, fills a volume V, each individual particle has zero volume. The gas fills a volume V because each molecule is in motion such that, given enough time, it explores the total extent of the volume available to it in a container. When the molecules collide with each other, or with the walls of the container, they exchange energy and momentum. There is no loss of energy when summed over the collision partners; hence, the collisions are elastic. There is conservation not only of energy but also of linear momentum. Because there are no intermolecular interactions, the only way in which ideal gas molecules influence each other is when they collide. Their energy and the energy of the system do not depend on how close or far apart the particles are from one another.
There are different types of gases based on what types of molecule are present. The particles may be atoms or molecules (we consider only neutral species here). A gas of atoms is also called a monatomic gas. The molecules can be classified by the number of atoms they have. We make three distinctions here. Diatomics contain two atoms, while triatomics and all gases with several atoms are polyatomic molecules. There are also ‘big’ molecules. Although ‘big’ is not well defined, what is meant here is polymers, proteins, DNA and other such molecules. These very large molecules can sometimes be put in the gas phase by laser desorption or electrospray techniques. Because these are special cases, we need not concern ourselves with big molecules in the gas phase for now. Rather, our discussion will revolve around monatomic, diatomic and polyatomic gases. The reason for dividing gases up along these lines will become apparent as soon as we consider how energy is partitioned in gases.
2.1 Ideal gas equation of state
We begin by discussing the properties of an ideal gas in the thermodynamic limit. The thermodynamic limit is defined as letting the volume approach infinity while maintaining a constant temperature (and chemical potential). More meaningfully for this discussion, it is the limit in which the size and shape of the container about the gas have no influence on its properties. There are many more molecules in the bulk of the gas than in contact with any surface, and the walls of the container are far apart compared to the size of the molecule. We shall see later that the influences of breaking such a limit are the hallmarks of nanoscience. An ideal gas obeys the ideal gas law

where p is the pressure, V the volume, n the number of moles, R the gas constant, and T the temperature. Equation (2.1) is also an equation of state. It tells us how the state of the gas changes as the conditions of the gas change. It is a mathematical expression of the experimental fact that, for a given amount of any fluid (n moles of gas or liquid, ideal or not), any two of the variables p, V, and T constitute a complete set of independent variables. In other words, all fluids regardless of their microscopic structure have an equation of state f(p, V, T) = 0. As we shall see shortly, T is a proxy variable for molecular energy. Therefore, fluids other than ideal ones, which possess intermolecular interactions that influence their energy, will have different equations of state. Deviations from the ideal gas law will be greatest for those species that interact most strongly.
The ideal gas law can be derived from statistical mechanics. However, it can also be derived empirically and independently by developing an equation that is consistent with experimental results. The following experimental observations have been made in the limit of low pressure (the dilute gas limit), that is, they hold for ideal gases:
Boyle’s law

Charles’s/Gay-Lussac’s law


which implies

Avogadro’s principle states that

Therefore, we can construct an equation, the ideal gas law Eq (2.1), which is consistent with all of these conditions. The constant that makes the ideal gas law consistent is R, the (universal) gas constant. R is determined by finding the value of the following expression in the limit of low pressure

Note that the Boltzmann constant is defined as the gas constant per molecule; thus, division by Avogadro’s constant yields its value,

and the ideal gas law can be written either on a per mole or per molecule basis

Once we have the ideal gas law, the laws of Boyle and Charles and Gay-Lussac are superfluous as they can all be derived from pV = nRT. In other words, it is a good rule to learn the most fundamental equation and then manipulate it to derive other useful results. As an example, introducing the mass density ρ defined by
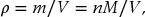
where m is the molecular mass, M the molar mass and V the volume, we can rewrite the ideal gas law as

2.1.1 Example
At 500 °C and 93.2 kPa, the mass density of sulfur vapor is 3.710 kg m−3. What is the molecular formula of sulfur under these circumstances?
Use the ideal gas law and the definition of density. First, rearrange the definition of density ρ

to arrive at an expression for the molar mass M,

Then rearrange the ideal gas law to obtain an expression for V/n
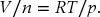
Now substitute the expression for V/n into the expression for M

The atomic weight of S is 32 g mol−1; 256/32 = 8, so therefore the molecular formula is S8.
2.1.2 Directed practice
Assuming that the molar mass is a constant, derive an expression for the density of an ideal gas from the ideal gas law as a function of temperature. Why might this formula break down for sulfur at temperatures much above 500 °C?
[Answer: ρ = pM/RT]
We should at this point pause and back up. To understand why an ideal gas follows the equation of state given above, we need to define the terms in it and see how these relate to molecular energy. In other words, we need to demonstrate what we really mean by pressure and define a scale for absolute temperature.
2.2 Molecular degrees of freedom
All gas-phase molecules in a macroscopic container, regardless of whether they are monatomic, diatomic or polyatomic, can translate in three directions. In other words, they have three translational degrees of freedom. In macroscopic containers at temperatures of interest to any form of usual chemistry, the translational degrees of freedom are completely classical. By that we mean that they are accurately described by the laws of classical mechanics. In principle, they can also possess electronic excitations, although electronic states are generally separated by large differences in energy. Usually, molecules are in their ground electronic state (the ground state is the lowest energy state). Only at very high temperatures are a significant number of molecules in electronic excited states (excited states are states that are at higher energy than the ground state), so we need not consider them further here. Similarly, we will only consider nuclear degrees of freedom when we deem them necessary, and will not discuss them in this general case. For a monatomic gas that is the end of the story as far as degrees of freedom are concerned.
Diatomic and polyatomic molecules possess bonds between atoms. Molecules are free to rotate in space and the bonds can vibrate. Each atom has three degrees of freedom before it enters the molecule. A molecule with N atoms must therefore have 3N degrees of freedom. Three of these degrees are translational, while the remaining (3N − 3) degrees correspond to internal degrees of freedom of rotation and vibration. A diatomic molecule can only vibrate in one way, in that the bond can stretch and contract. The two remaining degrees are rotational degrees of freedom. Think of a pencil. You can rotate it in the plane of the table or perpendicular to the plane of the table. Any other orientation of rotation can be constructed as a combination of in-plane and out-of-plane components. Rotation along the internuclear axis is of no consequence. The same partitioning of three translations and two rotations holds for any linear polyatomic molecule, which leaves (3N − 5) vibrational degrees of freedom. A nonlinear polyatomic molecule has three translational degrees as well as three rotational degrees of freedom (x, y, and z components to both translational and rotational motion). The remaining (3N − 6) degrees are vibrational degrees of freedom. As the molecular size increases, the number of available vibrational states increases rapidly. As a first approximation, we will consider that the degrees of freedom are independent of each other.
Rotations and vibrations are inherently quantum mechanical on a molecular level. We will explore this in detail in later chapters, but for now it will suffice to describe the broad details of the models we use to describe rotating and vibrating molecules.
The first-order approximation to a rotating diatomic molecule is a rigid rotor (Fig. 2.1). A rigid rotor is composed of a massless rod of fixed length corresponding to the equilibrium bond distance Re. This rod connects two point masses m1 and m2. Quantum mechanically, the rigid rotor is only allowed to attain certain values of energy; that is, the rotational energy level structure is quantized. So, why is rotation quantized but translation is not? Translation occurs on the distance scale of the container in which the molecule resides. As long as this container is large compared to the molecule, translation acts classically and has a continuous distribution of energy levels. As we shall see later, we can model rotation as the motion of a particle on a sphere (or spheroid) of molecular dimensions. Rotation is confined to a distance of molecular dimensions and is, therefore, quantized. Confinement to small dimensions is inherent for bringing out quantized behavior.

Figure 2.1 (a) Rigid rotor with rotational axis J and equilibrium bond distance Re between atoms of mass m1 and m2. (b) The corresponding quantized energy level structure for states with rotational quantum number 0 ≤ J ≤ 14.
The energy level spacing is larger for lighter masses and shorter bonds. Thus, H2 has much larger energy spacings than I2. The energy of a rigid rotor is

with a rotational constant B defined by

where J = 0, 1, 2, … is the rotational quantum number, h is Planck’s constant, ℏ = h/2π (pronounced h bar and called the reduced Planck constant), and c is the speed of light. (B is conventionally given in cm−1 rather than the SI unit of m−1.) By using a value of c = 3.00 × 1010 cm s− 1, the unit cm is cancelled in Eq. (2.12) and the energy is given in J. Note that J only takes on discrete values as expected for a quantized variable, rather than a continuous distribution of values as would be expected for a classical variable. The reduced mass is defined by

The energy level spacing between two successive levels is given by

Since B = 0.03737 cm−1 and 60.85 cm−1 for I2 and H2, respectively, these spacings are on the order of 0.1–100 cm−1 = 0.012–1.2 kJ mol−1 = 0.12–12 meV per molecule. This compares to kBT = 25 meV at room temperature. Thus, at sufficiently high temperatures many rotational levels are populated in heavy molecules, and the distribution of molecules that populate these states is approximated by a classical continuous distribution. Diatomic hydrides and lighter molecules have significantly fewer, discretely populated levels (we will discuss this distribution in detail later). Equation (2.15) shows that the energy level spacing increases with increasing J. However, because space is also quantized (or at least the direction into which the rotational angular momentum is quantized), each level contains (2J + 1) levels at the same energy (corresponding to the (2J + 1) directions into which the rotational angular momentum vector can point). We say that each level has a degeneracy of (2J + 1) and that levels at the same energy are degenerate. Since both energy level spacing and degeneracy are increasing functions of J, we can ask how the density of states increases – that is, how does the number of states per unit energy change? The density of states ρ(J) is defined (in the limit of large J) by

Equation (2.16) shows that the rotational density of state is constant.
2.2.1 Directed practice
Perform the conversions of energy units to show that 0.1–100 cm−1 = 0.012–1.2 kJ mol−1 = 0.12–12 meV.
The harmonic oscillator (Fig. 2.2) is the first approximation for the vibrational motion of a bond. The harmonic oscillator energy levels are quantized according to
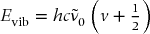
where the fundamental wavenumber  (again in cm−1) depends on the force constant k and reduced mass μ according to
(again in cm−1) depends on the force constant k and reduced mass μ according to


Figure 2.2 (a) An oscillator (i.e., a molecule) with atoms of mass m1 and m2 connected by a spring of force constant k at an equilibrium bond distance Re. The energy level structure of a quantum mechanical (b) harmonic and (c) Morse oscillator. The Morse oscillator shown corresponds to the H2 molecule. The rotational level spacings are much smaller in energy than the vibrational levels.
The vibrational quantum number v again takes on integer values v = 0, 1, 2…. Larger values of k and  correlate with stiffer, stronger bonds. Again, H2 with the highest fundamental wavenumber
correlate with stiffer, stronger bonds. Again, H2 with the highest fundamental wavenumber  is anomalously high. I2 exhibits a fundamental wavenumber of only 215 cm−1. Bonds tend to be harmonic in the ground state, but at higher and higher levels of excitation the energy levels are spaced progressively closer together and eventually the bond breaks. In other words, the density of states of a real oscillator increases with increasing vibrational energy. These aspects of anharmonicity are better represented by the Morse oscillator energy level structure shown in Fig. 2.2(c). To a first approximation, we consider each vibrational degree of freedom as corresponding to its own independent oscillator.
is anomalously high. I2 exhibits a fundamental wavenumber of only 215 cm−1. Bonds tend to be harmonic in the ground state, but at higher and higher levels of excitation the energy levels are spaced progressively closer together and eventually the bond breaks. In other words, the density of states of a real oscillator increases with increasing vibrational energy. These aspects of anharmonicity are better represented by the Morse oscillator energy level structure shown in Fig. 2.2(c). To a first approximation, we consider each vibrational degree of freedom as corresponding to its own independent oscillator.
Analogous to the rotational energy distribution, there is a high-temperature limit in which vibrational energy will be well approximated by a classical continuous distribution. This is most likely to be reached for polyatomic molecules containing heavy atoms. Since vibrational energy spacings are typically much larger than rotational energy level spacings, quantum mechanical corrections to classical expectations are much more likely when vibrations need to be considered.
2.3 Translational energy: Distribution and relation to pressure
The kinetic hypothesis states that the molecules that make up matter are constantly in motion, even when body of matter is at rest. Here, we consider the translational motion of an ideal gas, also known as a perfect gas. That molecules are translating means that they have both kinetic energy ( ) and linear momentum (mv), both of which depend on the molecular mass m and its velocity v. Momentum-changing collisions of gas molecules with the container walls cause a force to be exerted on the walls. Pressure is a measure of this force per unit area. Thus, the mass and velocity of the molecules contribute to both the internal energy of the sample and its pressure. To simplify matters further we will assume that our perfect gas is not only colliding elastically (no internal energy changes including electronic excitation) and composed of noninteracting particles, but also that it is dilute (far from condensing) and composed of monatomic particles.
) and linear momentum (mv), both of which depend on the molecular mass m and its velocity v. Momentum-changing collisions of gas molecules with the container walls cause a force to be exerted on the walls. Pressure is a measure of this force per unit area. Thus, the mass and velocity of the molecules contribute to both the internal energy of the sample and its pressure. To simplify matters further we will assume that our perfect gas is not only colliding elastically (no internal energy changes including electronic excitation) and composed of noninteracting particles, but also that it is dilute (far from condensing) and composed of monatomic particles.
Consider a gas that is isolated from the rest of the universe (no exchange of energy or matter) such that the macroscopic properties are not changing over time. We state that this system is ‘at equilibrium.’ When averaged over time, its temperature (which we have yet to define) and pressure are constant. Both of these quantities are dependent on the velocity distribution of the gas molecules; therefore, the time average of the velocity distribution must also be constant. This does not mean that the velocity of each individual gas molecule is constant. On the contrary, the molecules are continually colliding with each other and exchanging momentum. However, because there is a huge number of molecules, say, on the order of Avogadro’s number, the individual fluctuations experienced by each molecule average out. There will be fluctuations in the velocity distribution, but the magnitude of these fluctuations tends to be small compared to the mean value averaged over the whole distribution. This in turn means that the fluctuations in p and T are small compared to their mean values. At equilibrium, the gas is also uniformly distributed throughout the container and the container is sufficiently large such that any property measured is independent of the shape of the container or the position in the container at which a measurement is performed. Thus, for instance, Pascal’s law – that the pressure is equal in all directions – holds at equilibrium.
The distribution of molecular velocities and positions is not only homogeneous (uniform in space) but also isotropic (uniform in all directions) at equilibrium. This means that there are no net flows in the gas, which requires that the average (indicated by angle brackets) of velocity vectors must vanish

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


