The term pituitary (derived from both Latin and Greek) literally means to “spit mucus,” reflecting the primitive notion of pituitary function. In one way, the ancient physiologists were correct—they believed that the brain was responsible for signaling the pituitary to secrete; however, instead of mucus, it was later discovered that the brain directs the pituitary to secrete hormones that regulate other endocrine glands. When this was recognized, the pituitary was designated the “master gland” because, without the pituitary, there was a cessation of growth, together with profound alterations in intermediary metabolism and failure of gonadal, thyroidal, and adrenal function. The pituitary is also referred to as the hypophysis, from Greek meaning “undergrowth,” attesting to its unique position under the hypothalamus.
Our concept of pituitary function and its role in regulating other endocrine glands has changed. Rather than viewed as the master gland, it is more appropriately recognized as a transponder that translates neural input into a hormonal product. Features that distinguish the function of the pituitary include feedback loops, pulsatile secretions, diurnal rhythms, and environmental or external modification of its performance. These characteristics of pituitary operation can vex the clinical evaluation of suspected endocrine disease or, alternatively, lend incredible insight into subtle defects in endocrinologic function.
EMBRYOLOGY AND ANATOMY
The three distinct parts of the pituitary are the anterior pituitary, or adenohypophysis; the intermediate lobe, or pars intermedialis; and the posterior pituitary, or neurohypophysis. The intermediate lobe is poorly developed in humans and has little functional capacity other than to confuse radiologists by forming nonfunctional, benign, cystic enlargements of the pituitary. The posterior pituitary, which arises from the diencephalon, is responsible for the storage and release of oxytocin and vasopressin (also called antidiuretic hormone [ADH]). The anterior pituitary, the largest portion of the gland, originates from Rathke pouch, an evagination of buccal ectoderm that progressively extends upward and is eventually enveloped by the sphenoid bone. The creation of the median eminence, the inferior portion of the hypothalamus, and the pituitary stalk is the other critical event in the formation of the hypothalamic–hypophyseal unit. Pituitary function can be detected between the seventh and ninth weeks of gestation. The ultimate determination of anterior pituitary cell types is dependent on the spatial relationships of progenitor cell types and an integrated flow of transcription factors that ultimately form lactotrophs (prolactin-secreting cells), somatotrophs (growth hormone [GH]-secreting cells), thyrotrophs (thyroid-stimulating hormone [TSH]-secreting cells), corticotrophs (adrenocorticotropin hormone [ACTH]-secreting cells), and gonadotrophs (luteinizing hormone [LH]- and follicle-stimulating hormone [FSH]-secreting cells).1
The pituitary resides in a pocket of the sphenoid (the sella turcica, meaning “Turkish saddle”) and is surrounded by dura mater. The reflection of dura that separates the superior portion of the pituitary from the hypothalamus, the diaphragma sella, is penetrated by the infundibulum, or pituitary stalk, that connects the adenohypophysis to the median eminence and hypothalamus. The pituitary stalk contains both neural and vascular structures that terminate in the hypophysis. The posterior pituitary is connected to the supraoptic and paraventricular hypothalamic nuclei (where vasopressin and oxytocin are produced) by way of two, distinct neurosecretory tracts, the supraopticohypophyseal and tuberohypophyseal tracts, which pass through the stalk. The anterior pituitary receives 80% to 90% of its blood supply and many hypothalamic factors via the hypothalamic–hypophyseal portal system, also contained in the stalk. The primary plexus of this portal system is located in the median eminence and is composed of capillaries lacking a blood–brain barrier (fenestrated capillaries), where the hypothalamic nuclei that modulate pituitary function terminate their axons. In turn, the long portal vessels connect the primary plexus to the anterior pituitary and serve as a conduit for these hypothalamic–hypophysiotropic hormones. These anatomic relationships are illustrated in Figure 20.1.
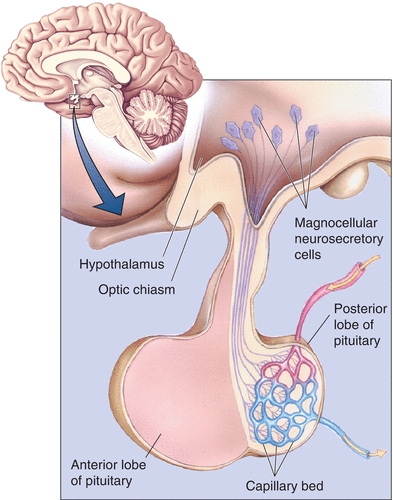
FIGURE 20.1 Relational anatomy of the pituitary and hypothalamus. (Reproduced with permission from Bear MF, Connors BW, Paradiso MA. Neuroscience: Exploring the Brain. 2nd ed. Baltimore, MD: Lippincott Williams & Wilkins; 2001:501.)
FUNCTIONAL ASPECTS OF THE HYPOTHALAMIC–HYPOPHYSEAL UNIT
Afferent pathways (inputs) to the hypothalamus are integrated in various specialized nuclei, processed, and then resolved into specific patterned responses. Because the hypothalamus has many efferent neural connections (outputs) to higher brain centers (the limbic system, the autonomic nervous system, and the pituitary), these responses appear to be rather diffuse but are actually stereotypical. The hypothalamic response patterns are similar for each specific pituitary hormone and characterized by open-loop negative feedback mechanisms, pulsatility, and cyclicity. Negative feedback resembles a typical servomechanism and forms the basis of our understanding of hypothalamic–pituitary function. An example of negative feedback is the relationship between a thermostat and a home heating unit. The thermostat is set to a given temperature. As the temperature in the home falls below this set point, the thermostat sends an electrical impulse to the furnace and turns the furnace on. Heat is restored to the room, and, when the temperature in the room exceeds the predetermined set point, the thermostat turns off the furnace. Because the thermostat set point can be adjusted for the comfort of the occupants, the furnace–thermostat functional relationship is termed an open-loop negative feedback system. Most endocrine feedback loops are of the open-loop variety, meaning that they are subject to external modulation and generally influenced or modified by higher neural input or other hormones.
A simple example of an endocrine feedback loop is the hypothalamic–pituitary–thyroidal axis. The hypothalamus produces the hypophysiotropic hormone, thyrotropin-releasing hormone (TRH), and releases it into the portal system where it directs the thyrotrophs (or TSH-producing cells) in the anterior pituitary to secrete TSH. TSH circulates to the thyroid and stimulates several steps in the thyroid that are critical in the production and release of thyroid hormone (thyroxine). Thyroxine is released in the blood and circulates to the hypothalamus and pituitary to suppress further TRH and TSH production. This axis can be partially inhibited by adrenal steroids (glucocorticoids) and by cytokines; as a result, thyroid hormone production may decline during periods of severe physiologic stress.2 The feedback of thyroxine at the level of the pituitary is called a short feedback loop, and feedback at the level of the hypothalamus is called a long feedback loop. Feedback between the pituitary and hypothalamus (when present) is called an ultrashort feedback loop. Figure 20.2 illustrates this simple feedback loop.
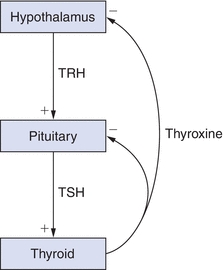
FIGURE 20.2 Simple feedback loop. TRH, thyrotropin-releasing hormone; TSH, thyroid-stimulating hormone.
All anterior pituitary hormones are secreted in a pulsatile fashion. The pulse frequency of secretion is generally regulated by neural modulation and is specific for each hypothalamic–pituitary–end-organ unit. Perhaps the best example of pituitary pulsatility is the secretion of the hormones that regulate gonadal function (LH and FSH). In normal male subjects, the median interpulse interval for LH is 55 minutes, and the average LH peak duration is 40 minutes.3 The pulse frequency of the regulatory hypothalamic hormone, gonadotropin-releasing hormone (GnRH), has profound effects on LH secretion profiles—increasing the frequency of GnRH pulses reduces the gonadotrope secretory response, and decreasing the GnRH pulse frequency increases the amplitude of the subsequent LH pulse.4
Another feature of the hypothalamic–pituitary unit is the cyclic nature of hormone secretion. The nervous system usually regulates this function through external signals, such as light–dark changes or the ratio of daylight to darkness. The term zeitgeber (“time giver”) refers to the process of entraining or synchronizing these external cues into the function of internal biologic clocks. As a result, many pituitary hormones are secreted in different amounts, depending on the time of day. These circadian, or diurnal, rhythms are typified by ACTH, or TSH secretion. With ACTH, the nadir of secretion is between 11:00 pm and 3:00 am, and the peak occurs on awakening or around 6:00 to 9:00 am.5 The circadian rhythm of ACTH is a result of variations in pulse amplitude and not alterations in pulse frequency.6 The nocturnal levels of TSH are approximately twice the daytime levels; the nocturnal rise in TSH is a result of increased pulse amplitude.7
HYPOPHYSIOTROPIC OR HYPOTHALAMIC HORMONES
The hypothalamus produces many different products; however, only those that have a direct effect on classic pituitary function will be discussed in this chapter. Most products are peptides; however, bioactive amines are also synthesized and transported from the hypothalamus. Hypothalamic hormones may have multiple actions. For example, TRH stimulates the secretion of both TSH and prolactin; GnRH stimulates both LH and FSH production; and somatostatin (SS) inhibits GH and TSH release from the pituitary. In addition to its effects on water metabolism, vasopressin (ADH) can also stimulate ACTH secretion. The main stimulus for ACTH secretion is corticotropin-releasing hormone. These hypophysiotropic hormones are found throughout the central nervous system and various other tissues, including the gut, pancreas, and other endocrine glands. Their function outside the hypothalamus and pituitary is poorly understood. The action of hypophysiotropic hormones on anterior pituitary function is summarized in Table 20.1.
TABLE 20.1 Hypophysiotropic Hormones
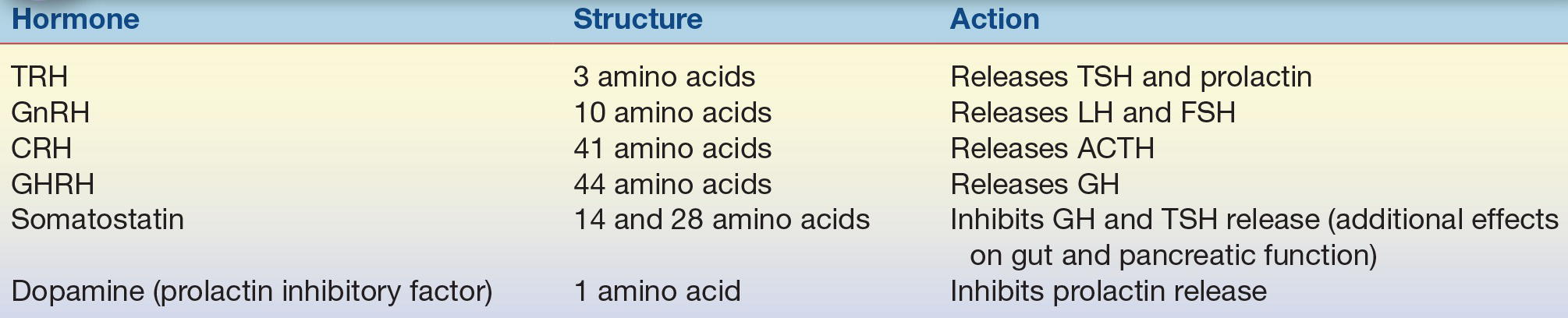
TRH, thyrotropin-releasing hormone; TSH, thyroid-stimulating hormone; GnRH, gonadotropin-releasing hormone; LH, luteinizing hormone; FSH, follicle-stimulating hormone; CRH, corticotropin-releasing hormone; ACTH, adrenocorticotropin hormone; GHRH, growth hormone–releasing hormone.
ANTERIOR PITUITARY HORMONES
The hormones secreted from the anterior pituitary are larger and more complex than those synthesized in the hypothalamus. These pituitary hormones are either tropic, meaning their actions are specific for another endocrine gland, or they are direct effectors, because they act directly on peripheral tissue. TSH and its unique role in regulating thyroid function provide an example of tropic; an example of a direct effector is GH. GH has direct effects on substrate metabolism in numerous tissues and also stimulates the liver to produce growth factors that are critical in enhancing linear growth. The tropic hormones are LH, which directs testosterone production from Leydig cells in men and ovulation in women; FSH, which is responsible for ovarian recruitment and early folliculogenesis in women and spermatogenesis in men; TSH, which directs thyroid hormone production from the thyroid; and ACTH, which regulates adrenal steroidogenesis. Both GH and prolactin are direct effectors. A general summary of relationships among anterior pituitary hormones and their target organs and feedback effectors is provided in Table 20.2.
TABLE 20.2 Anterior Pituitary Hormones

LH, luteinizing hormone; FSH, follicle-stimulating hormone; TSH, thyroid-stimulating hormone; ACTH, adrenocorticotropin hormone; T4, thyroxine; T3, triiodothyronine; E2, estradiol; T, testosterone; POMC, pro-opiomelanocortin; IGF-I, insulin-like growth factor.
The actions of the tropic hormones are discussed in other chapters devoted to the specific target gland.
PITUITARY TUMORS
According to autopsy studies, up to 20% of people harbor clinically silent pituitary adenomas, and findings consistent with pituitary tumors are observed in 10% to 30% of normal individuals undergoing MRI examinations. In addition, pituitary tumors account for 91% of the lesions removed from carefully selected patients who have undergone transsphenoidal surgery.8,9 Close medical follow-up is recommended if the incidentally discovered lesion is hormonally silent and is less than 1 cm in diameter. Prolactin-secreting pituitary tumors are the most common, followed by nonfunctioning or null cell tumors, and tumors that secrete GH, gonadotropins, ACTH, or TSH account for the remainder. The WHO defined “atypical pituitary tumors” as tumors that have an MIB-1 proliferative index greater than 3%, excessive p53 immunoreactivity, and increased mitotic activity.10 Most of these “atypical” tumors are macroadenomas (i.e., >1 cm in diameter) and show invasion into surrounding structures like the cavernous sinuses. They may or may not be hormonally active and, interestingly, tend to stain commonly for GH or ACTH even though they may not produce clinically evident syndromes.10 MIB-1 is a monoclonal antibody that is used to detect the Ki-67 antigen, a marker of cell proliferation, and a high “proliferation index” suggests higher degree of atypia.11
Physiologic enlargement of the pituitary can be seen during puberty and pregnancy. The enlargement seen during pregnancy is due to lactotroph hyperplasia. Thyrotroph and lactotroph or gonadotroph hyperplasia can also be seen in long-standing primary thyroidal or gonadal failure, respectively.
GROWTH HORMONE
The pituitary is vital for normal growth. Growth ceases if the pituitary is removed, and if the hormonal products from other endocrine glands that are acted on by the pituitary are replaced (thyroxine, adrenal steroids, and gonadal steroids), growth is not restored until GH is administered. However, if GH is given in isolation without the other hormones, growth is not promoted. Therefore, it takes complete functioning of the pituitary to establish conditions ripe for growth of the individual. It also takes adequate nutrition, normal levels of insulin, and overall good health to achieve a person’s genetic growth potential.
GH, also called somatotropin, is structurally related to prolactin and human placental lactogen. A single peptide with two intramolecular disulfide bridges, it belongs to the direct effector class of anterior pituitary hormones. The somatotrophs, pituitary cells that produce GH, constitute over one-third of normal pituitary weight. Release of somatotropin from the pituitary is stimulated by the hypothalamic peptide growth hormone–releasing hormone (GHRH); somatotropin’s secretion is inhibited by SS.12 GH is secreted in pulses, with an average interpulse interval of 2 to 3 hours, with the most reproducible peak occurring at the onset of sleep.13 Between these pulses, the level of GH may fall below the detectable limit, resulting in the clinical evaluation of GH deficiency being based on a single, challenging measurement. Ghrelin, an enteric hormone that plays important roles in nutrient sensing, appetite and in glucose regulation, is also a potent stimulator of GH secretion.14
No other hypothalamic–hypophyseal system more vividly illustrates the concept of an open-loop paradigm than that seen with GH. The on-and-off functions of GHRH/SS and the basic pattern of secretory pulses of GH are heavily modulated by other factors (Table 20.3).
TABLE 20.3 Other Modifiers of Growth Hormone Secretion

Actions of GH
GH has many diverse effects on metabolism; it is considered an amphibolic hormone because it directly influences both anabolic and catabolic processes. One major effect of GH is that it allows an individual to effectively transition from a fed state to a fasting state without experiencing a shortage of substrates required for normal intracellular oxidation. GH directly antagonizes the effect of insulin on glucose metabolism, promotes hepatic gluconeogenesis, and stimulates lipolysis.15,16 From a teleologic viewpoint, this makes perfect sense—enhanced lipolysis provides oxidative substrate for peripheral tissue, such as skeletal muscle, and yet conserves glucose for the central nervous system by stimulating the hepatic delivery of glucose and opposing insulin-mediated glucose disposal. Indeed, isolated GH deficiency in children may be accompanied by hypoglycemia; however, hypoglycemia is more likely to occur if both GH and ACTH are deficient.17
The anabolic effects of GH are reflected by enhanced protein synthesis in skeletal muscle and other tissues. This is translated into a positive nitrogen balance and phosphate retention.
Although GH has direct effects on many tissues, it also has indirect effects that are mediated by factors that were initially called somatomedins. In early experiments, it became apparent that GH supplementation in hypophysectomized animals induced the production of an additional factor that stimulated the incorporation of sulfate into cartilage.18,19 As this “protein” was purified, it was evident that there was more than one somatomedin, and because of their structural homology to proinsulin, the nomenclature shifted to insulin-like growth factor (IGF).20,21 For example, somatomedin C, the major growth factor induced by GH, is now IGF-I.22 IGFs also have cell surface receptors that are distinct from insulin; however, supraphysiologic levels of IGF-II can “bleed” over on the insulin receptor and cause hypoglycemia,23 and hyperinsulinemia can partially activate IGF-I receptors.24 GH stimulates the production of IGF-I from the liver, and as a result, IGF-I becomes a biologic amplifier of GH levels. IGFs are complexed to specific serum binding proteins that have been shown to affect the actions of IGFs in multifaceted ways.25 IGF-binding protein 3 (IGFBP-3) is perhaps the best studied member of the IGFBP family. Recently, IGFBPs and specifically IGFBP-3 have been shown to directly play a role in the pathophysiology of several human cancers (this may be independent of IGF-1 and IGF-1 receptor–mediated pathways).26 The tumor suppressor gene, P53, has been shown to upregulate active IGFBP-3 secretion, which in turn inhibits IGF-1 signaled mutagenesis, and, thus, inhibits neoplastic cell proliferation.27 Low levels of IGFBP-3 were positively correlated with higher rates of colorectal cancer risk (in men) in a nested case–control study from the Physicians Health Study cohort.28
Testing
As noted above, a single, random measurement of GH is rarely diagnostic. The current testing paradigms for GH are soundly based on the dynamic physiology of the GH axis. For example, circulating levels of IGF-I and, perhaps, IGFBP-3 reasonably integrate the peaks of GH secretion, and elevated levels of both are consistent with, but may not be diagnostic of, a sustained excess of GH. Other conditions such as hepatoma can be associated with high levels of IGF-I, and levels of IGFBP-3 may be inappropriately normal in some people with active acromegaly. Conversely, low IGF-I levels may reflect inadequate production of GH; however, low IGF levels are also seen in patients with poorly controlled diabetes, malnutrition, or other chronic illnesses.29 The inherent high biologic variability and assay performance issues further confound the use of IGF-I measurements in the clinical setting.30 Recently, new recommendations to improve the assay performance for both IGF-I and GH have been published.31
Definitive testing for determining the autonomous production of GH relies upon the normal suppressibility of GH by oral glucose loading.22,32,33 This test is performed after an overnight fast, and the patient is given a 100 g oral glucose load. GH is measured at time zero and at 60 and 120 minutes after glucose ingestion. Following oral glucose loading, GH levels are undetectable in normal individuals; however, in patients with acromegaly, GH levels fail to suppress and may even paradoxically rise.
Testing patients for suspected GH deficiency is more complicated. There are several strategies to stimulate GH, and new protocols are currently evolving. Once considered the gold standard, insulin-induced hypoglycemia is being replaced by less uncomfortable testing schemes.34 Combination infusions of GHRH and the amino acid l-arginine or an infusion of l-arginine coupled with oral l-DOPA are the most widely used although acquiring some of the medications can be problematic.35 If GH levels rise above 3 to 5 ng/mL, it is unlikely that the patient is GH deficient34; however, a lower threshold may be adopted because of improved sensitivity of the newer two-site GH assays.35 On the other hand, several studies have shown that provocative GH testing may not be necessary in patients with low IGF-1 levels and otherwise documented panhypopituitarism.29
Acromegaly
Acromegaly results from pathologic or autonomous GH excess and, in the vast majority of patients, is a result of a pituitary tumor. There have been isolated case reports of tumors causing acromegaly as a result of the ectopic production of GHRH,36, 37, 38 and although exceedingly interesting or instructive, the ectopic production of GHRH or GH remains rare.37 Recent reports have documented mutations in the aryl hydrocarbon–interacting protein gene (AIP)39 in cases of familial acromegaly and polymorphisms in the SS receptor type 5 gene in sporadic cases.40 If a GH-producing tumor occurs before epiphyseal closure, the patient develops gigantism41 and may grow to an impressive height; otherwise, the patient develops classical, but insidious, features of bony and soft tissue overgrowth.42 These features include progressive enlargement of the hands and feet as well as growth of facial bones, including the mandible and bones of the skull. In advanced cases, the patient may develop significant gaps between their teeth. Diffuse (not longitudinal if the condition occurred following puberty) overgrowth of the ends of long bones or the spine can produce a debilitating form of arthritis.43 Because GH is an insulin antagonist, glucose intolerance or overt diabetes can occur. Hypertension; accelerated atherosclerosis; and proximal muscle weakness, resulting from acquired myopathy,44 may be seen late in the illness. Sleep apnea is common. Organomegaly, especially thyromegaly, is common, but hyperthyroidism is exceedingly rare unless the tumor cosecretes TSH. GH excess is also a hypermetabolic condition, and as a result, acromegalic patients may complain of excessive sweating or heat intolerance. The features of acromegaly develop slowly over time, and the patient (or even their family) may be oblivious that changes in physiognomy have occurred. In these cases, the patient’s complaints may center on the local effects of the tumor (headache or visual complaints) or symptoms related to the loss of other anterior pituitary hormones (hypopituitarism). A careful, retrospective review of older photographs may be crucial in differentiating coarse features due to inheritance from the classical consequences of acromegaly. If left untreated, acromegaly shortens life expectancy because of increased risk of heart disease, resulting from the combination of hypertension, coronary artery disease, and diabetes/insulin resistance. Because patients with acromegaly also have a greater lifetime risk of developing cancer, cancer surveillance programs (especially regular colonoscopy) are recommended.22
CASE STUDY 20.1
A 48-year-old man seeks care for evaluation of muscle weakness, headaches, and excessive sweating. He has poorly controlled hypertension and, on questioning, admits to noticing a gradual increase in both glove and shoe size, as well as a reduction in libido. A review of older photographs of the man documents coarsening of facial features, progressive prognathism, and broadening of the nose. Acromegaly is suspected.
questions
1. What screening tests are available?
2. What is the definitive test for autonomous growth hormone production?
3. Because the patient complains of reduced libido, hypogonadism is suspected. What evaluation is appropriate?
4. How would your thinking change if she had galactorrhea but normal levels of prolactin?
Cosecretion of prolactin can be seen in up to 40% of patients with acromegaly.45 Only a few TSH/GH-secreting tumors have also been reported.46
Confirming the diagnosis of acromegaly is relatively easy; however, some patients with acromegaly have normal random levels of GH. An elevated level of GH that does not suppress normally with glucose loading equates to an easy diagnosis. In those patients with normal, but inappropriately sustained, random levels of GH, elevated levels of IGF-I are helpful; however, nonsuppressibility of GH to glucose loading is the definitive test.32,33
Treatment of acromegaly can be challenging. The goal of treatment is tumor ablation, with continued function of the remainder of the pituitary. Transsphenoidal adenomectomy is the procedure of choice.22 If normal GH levels and kinetics (normal suppressibility to glucose) are restored following surgery, the patient is likely cured. Unfortunately, GH-producing tumors may be too large or may invade into local structures that preclude complete surgical extirpation, and the patient is left with a smaller, but hormonally active, tumor. External beam or focused irradiation is frequently used at this point, but it may take several years before GH levels decline.47,48 In the interim, efforts are made to suppress GH. Three different classes of agents, SS analogs (octreotide, pasireotide, and lanreotide), dopaminergic agonists (cabergoline and bromocriptine), and GH receptor antagonists (pegvisomant) may be employed for GH suppression.49,50
GH Deficiency
GH deficiency occurs in both children and adults. In children, it may be genetic or it may be due to tumors, such as craniopharyngiomas. In adults, it is a result of structural or functional abnormalities of the pituitary (see the section on hypopituitarism in this chapter); however, a decline in GH production is an inevitable consequence of aging, and the significance of this phenomenon is poorly understood.51,52
Although GH deficiency in children is manifest by growth failure, not all patients with short stature have GH deficiency53 (see above). There have been several genetic defects identified in the GH axis. The most common type is a recessive mutation in the GHRH gene that causes a failure of GH secretion. A rarer mutation, loss of the GH gene itself, has also been observed. Mutations that result in GH insensitivity have also been reported. These mutations may involve the GH receptor, IGF-I biosynthesis, IGF-I receptors, or defects in GH signal transduction. As a result, patients with GH insensitivity do not respond normally to exogenously administered GH. Finally, structural lesions of the pituitary or hypothalamus may also cause GH deficiency and may be associated with other anterior pituitary hormone deficiencies.
An adult GH deficiency syndrome has been described in patients who have complete or even partial failure of the anterior pituitary. The symptoms of this syndrome are extremely vague and include social withdrawal, fatigue, loss of motivation, and a diminished feeling of well-being,54 but several studies have documented increased mortality in children who are GH deficient although this relationship is less clear in adults.55 Osteoporosis and alterations in body composition (i.e., reduced lean body mass) are frequent concomitants of adult GH deficiency.56
GH replacement therapy has become relatively simple with the advent of recombinant human GH.57 Currently, the cost of GH is the major limiting factor for replacement. GH has been employed by athletes as a performance-enhancing substance and as an aid in injury recovery; however, the effectiveness of GH for these purposes is controversial.58
PROLACTIN
Prolactin is structurally related to GH and human placental lactogen. Considered a stress hormone, it has vital functions in relationship to reproduction. Prolactin is classified as a direct effector hormone (as opposed to a tropic hormone) because it has diffuse target tissue and lacks a single endocrine end organ.
Prolactin is unique among the anterior pituitary hormones because its major mode of hypothalamic regulation is tonic inhibition rather than intermittent stimulation. Prolactin inhibitory factor (PIF) was once considered a polypeptide hormone capable of inhibiting prolactin secretion; dopamine, however, is the only neuroendocrine signal that inhibits prolactin and is now considered to be the elusive PIF. Any compound that affects dopaminergic activity in the median eminence of the hypothalamus will also alter prolactin secretion. Examples of medications that cause hyperprolactinemia include phenothiazines, butyrophenones, metoclopramide, reserpine, tricyclic antidepressants, α-methyldopa, and antipsychotics that antagonize the dopamine D2 receptor. Any disruption of the pituitary stalk (e.g., tumors, trauma, or inflammation) causes an elevation in prolactin as a result of interruption of the flow of dopamine from the hypothalamus to the lactotrophs, the pituitary prolactin-secreting cells. TRH directly stimulates prolactin secretion and increases in TRH (as seen in primary hypothyroidism) elevate prolactin levels.59 Estrogens also directly stimulate lactotrophs to synthesize prolactin. Pathologic stimulation of the neural suckling reflex is the likely explanation of hyperprolactinemia associated with chest wall injuries. Hyperprolactinemia may also be seen in renal failure and polycystic ovary syndrome. Physiologic stressors, such as exercise and seizures, also elevate prolactin. The feedback effector for prolactin is unknown. Although the primary regulation of prolactin secretions is tonic inhibition (e.g., dopamine), it is also regulated by several hormones, including GnRH, TRH, and vasoactive intestinal polypeptide. Stimulation of breasts, as in nursing, causes the release of prolactin-secreting hormones from the hypothalamus through a spinal reflex arc.
As mentioned, the physiologic effect of prolactin is lactation. The usual consequence of prolactin excess is hypogonadism, either by suppression of gonadotropin secretion from the pituitary or by inhibition of gonadotropin action at the gonad.60 The suppression of ovulation seen in lactating postpartum mothers is related to this phenomenon.
Prolactinoma
A prolactinoma is a pituitary tumor that directly secretes prolactin, and it represents the most common type of functional pituitary tumor. The clinical presentation of a patient with a prolactinoma depends on the age and gender of the patient and the size of the tumor. Premenopausal women most frequently complain of menstrual irregularity/amenorrhea, infertility, or galactorrhea; men or postmenopausal women generally present with symptoms of a pituitary mass, such as headaches or visual complaints. Occasionally, a man may present with reduced libido or complaints of erectile dysfunction. The reason(s) for the varied presentations of a prolactinoma are somewhat obscure but likely relate to the dramatic, noticeable alteration in menses or the abrupt onset of a breast discharge in younger women. By contrast, the decline in reproductive function in older patients may be overlooked as an inexorable consequence of “aging.” One recently recognized complication of prolactin-induced hypogonadism is osteoporosis.61
CASE STUDY 20.2
A 23-year-old woman has experienced recent onset of a spontaneous, bilateral breast discharge and gradual cessation of menses. She reports normal growth and development and has never been pregnant.
questions
1. What conditions could be causing her symptoms?
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


