PART 16: Endocrinology and Metabolism
SECTION 1 | ENDOCRINOLOGY |
399 | Approach to the Patient with Endocrine Disorders |
The management of endocrine disorders requires a broad understanding of intermediary metabolism, reproductive physiology, bone metabolism, and growth. Accordingly, the practice of endocrinology is intimately linked to a conceptual framework for understanding hormone secretion, hormone action, and principles of feedback control (Chap. 400e). The endocrine system is evaluated primarily by measuring hormone concentrations, arming the clinician with valuable diagnostic information. Most disorders of the endocrine system are amenable to effective treatment once the correct diagnosis is determined. Endocrine deficiency disorders are treated with physiologic hormone replacement; hormone excess conditions, which usually are caused by benign glandular adenomas, are managed by removing tumors surgically or reducing hormone levels medically.
SCOPE OF ENDOCRINOLOGY
The specialty of endocrinology encompasses the study of glands and the hormones they produce. The term endocrine was coined by Starling to contrast the actions of hormones secreted internally (endocrine) with those secreted externally (exocrine) or into a lumen, such as the gastrointestinal tract. The term hormone, derived from a Greek phrase meaning “to set in motion,” aptly describes the dynamic actions of hormones as they elicit cellular responses and regulate physiologic processes through feedback mechanisms.
Unlike many other specialties in medicine, it is not possible to define endocrinology strictly along anatomic lines. The classic endocrine glands—pituitary, thyroid, parathyroid, pancreatic islets, adrenals, and gonads—communicate broadly with other organs through the nervous system, hormones, cytokines, and growth factors. In addition to its traditional synaptic functions, the brain produces a vast array of peptide hormones, and this has led to the discipline of neuroendocrinology. Through the production of hypothalamic releasing factors, the central nervous system (CNS) exerts a major regulatory influence over pituitary hormone secretion (Chap. 401e). The peripheral nervous system stimulates the adrenal medulla. The immune and endocrine systems are also intimately intertwined. The adrenal hormone cortisol is a powerful immunosuppressant. Cytokines and interleukins (ILs) have profound effects on the functions of the pituitary, adrenal, thyroid, and gonads. Common endocrine diseases such as autoimmune thyroid disease and type 1 diabetes mellitus are caused by dysregulation of immune surveillance and tolerance. Less common diseases such as polyglandular failure, Addison’s disease, and lymphocytic hypophysitis also have an immunologic basis.
The interdigitation of endocrinology with physiologic processes in other specialties sometimes blurs the role of hormones. For example, hormones play an important role in maintenance of blood pressure, intravascular volume, and peripheral resistance in the cardiovascular system. Vasoactive substances such as catecholamines, angiotensin II, endothelin, and nitric oxide are involved in dynamic changes of vascular tone in addition to their multiple roles in other tissues. The heart is the principal source of atrial natriuretic peptide, which acts in classic endocrine fashion to induce natriuresis at a distant target organ (the kidney). Erythropoietin, a traditional circulating hormone, is made in the kidney and stimulates erythropoiesis in bone marrow (Chap. 77). The kidney is also integrally involved in the renin-angiotensin axis (Chap. 406) and is a primary target of several hormones, including parathyroid hormone (PTH), mineralocorticoids, and vasopressin. The gastrointestinal tract produces a surprising number of peptide hormones, such as cholecystokinin, ghrelin, gastrin, secretin, and vasoactive intestinal peptide, among many others. Carcinoid and islet tumors can secrete excessive amounts of these hormones, leading to specific clinical syndromes (Chap. 113). Many of these gastrointestinal hormones are also produced in the CNS, where their functions are poorly understood. Adipose tissue produces leptin, which acts centrally to control appetite, along with adiponectin, resistin, and other hormones that regulate metabolism. As hormones such as inhibin, ghrelin, and leptin are discovered, they become integrated into the science and practice of medicine on the basis of their functional roles rather than their tissues of origin.
Characterization of hormone receptors frequently reveals unexpected relationships to factors in nonendocrine disciplines. The growth hormone (GH) and leptin receptors, for example, are members of the cytokine receptor family. The G protein–coupled receptors (GPCRs), which mediate the actions of many peptide hormones, are used in numerous physiologic processes, including vision, smell, and neurotransmission.
PATHOLOGIC MECHANISMS OF ENDOCRINE DISEASE
Endocrine diseases can be divided into three major types of conditions: (1) hormone excess, (2) hormone deficiency, and (3) hormone resistance (Table 399-1).
CAUSES OF ENDOCRINE DYSFUNCTION |
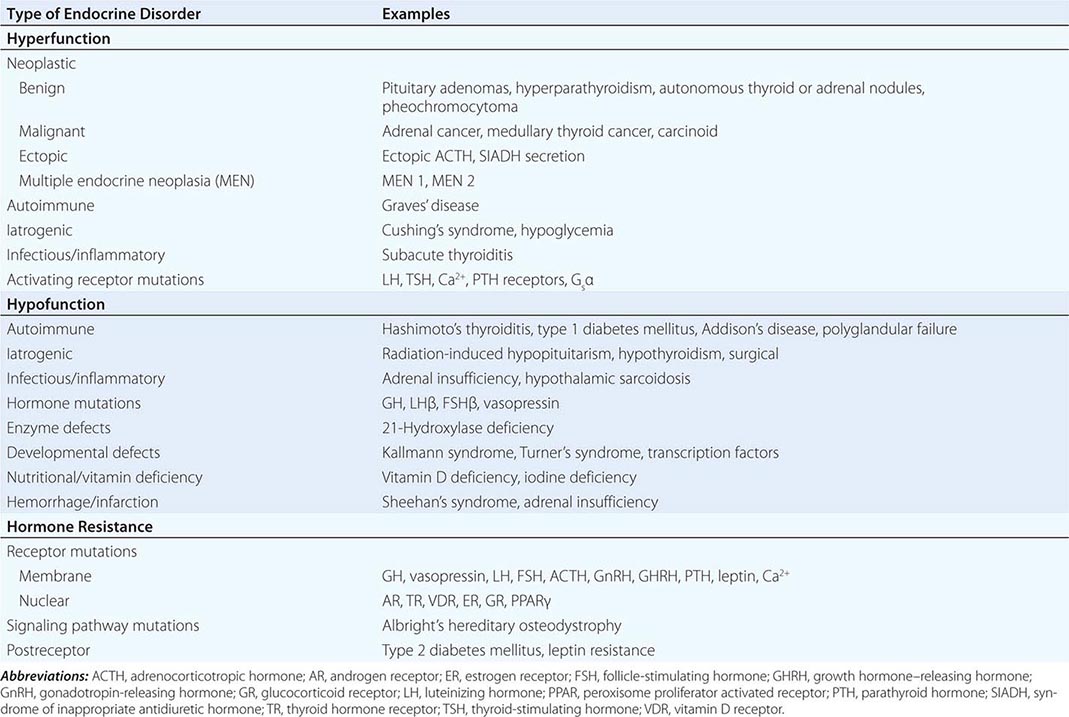
CAUSES OF HORMONE EXCESS
Syndromes of hormone excess can be caused by neoplastic growth of endocrine cells, autoimmune disorders, and excess hormone administration. Benign endocrine tumors, including parathyroid, pituitary, and adrenal adenomas, often retain the capacity to produce hormones, perhaps reflecting the fact that these tumors are relatively well differentiated. Many endocrine tumors exhibit subtle defects in their “set points” for feedback regulation. For example, in Cushing’s disease, impaired feedback inhibition of adrenocorticotropic hormone (ACTH) secretion is associated with autonomous function. However, the tumor cells are not completely resistant to feedback, as evidenced by ACTH suppression by higher doses of dexamethasone (e.g., high-dose dexamethasone test) (Chap. 406). Similar set point defects are also typical of parathyroid adenomas and autonomously functioning thyroid nodules.
The molecular basis of some endocrine tumors, such as the multiple endocrine neoplasia (MEN) syndromes (MEN 1, 2A, 2B), have provided important insights into tumorigenesis (Chap. 408). MEN 1 is characterized primarily by the triad of parathyroid, pancreatic islet, and pituitary tumors. MEN 2 predisposes to medullary thyroid carcinoma, pheochromocytoma, and hyperparathyroidism. The MEN1 gene, located on chromosome 11q13, encodes a putative tumor-suppressor gene, menin. Analogous to the paradigm first described for retinoblastoma, the affected individual inherits a mutant copy of the MEN1 gene, and tumorigenesis ensues after a somatic “second hit” leads to loss of function of the normal MEN1 gene (through deletion or point mutations).
In contrast to inactivation of a tumor-suppressor gene, as occurs in MEN 1 and most other inherited cancer syndromes, MEN 2 is caused by activating mutations in a single allele. In this case, activating mutations of the RET protooncogene, which encodes a receptor tyrosine kinase, leads to thyroid C cell hyperplasia in childhood before the development of medullary thyroid carcinoma. Elucidation of this pathogenic mechanism has allowed early genetic screening for RET mutations in individuals at risk for MEN 2, permitting identification of those who may benefit from prophylactic thyroidectomy and biochemical screening for pheochromocytoma and hyperparathyroidism.
Mutations that activate hormone receptor signaling have been identified in several GPCRs. For example, activating mutations of the luteinizing hormone (LH) receptor cause a dominantly transmitted form of male-limited precocious puberty, reflecting premature stimulation of testosterone synthesis in Leydig cells (Chap. 411). Activating mutations in these GPCRs are located predominantly in the transmembrane domains and induce receptor coupling to Gsα even in the absence of hormone. Consequently, adenylate cyclase is activated, and cyclic adenosine monophosphate (AMP) levels increase in a manner that mimics hormone action. A similar phenomenon results from activating mutations in Gsα. When these mutations occur early in development, they cause McCune-Albright syndrome. When they occur only in somatotropes, the activating Gsα mutations cause GH-secreting tumors and acromegaly (Chap. 403).
In autoimmune Graves’ disease, antibody interactions with the thyroid-stimulating hormone (TSH) receptor mimic TSH action, leading to hormone overproduction (Chap. 405). Analogous to the effects of activating mutations of the TSH receptor, these stimulating autoantibodies induce conformational changes that release the receptor from a constrained state, thereby triggering receptor coupling to G proteins.
CAUSES OF HORMONE DEFICIENCY
Most examples of hormone deficiency states can be attributed to glandular destruction caused by autoimmunity, surgery, infection, inflammation, infarction, hemorrhage, or tumor infiltration (Table 399-1). Autoimmune damage to the thyroid gland (Hashimoto’s thyroiditis) and pancreatic islet β cells (type 1 diabetes mellitus) is a prevalent cause of endocrine disease. Mutations in a number of hormones, hormone receptors, transcription factors, enzymes, and channels can also lead to hormone deficiencies.
HORMONE RESISTANCE
Most severe hormone resistance syndromes are due to inherited defects in membrane receptors, nuclear receptors, or the pathways that transduce receptor signals. These disorders are characterized by defective hormone action despite the presence of increased hormone levels. In complete androgen resistance, for example, mutations in the androgen receptor result in a female phenotypic appearance in genetic (XY) males, even though LH and testosterone levels are increased (Chap. 408). In addition to these relatively rare genetic disorders, more common acquired forms of functional hormone resistance include insulin resistance in type 2 diabetes mellitus, leptin resistance in obesity, and GH resistance in catabolic states. The pathogenesis of functional resistance involves receptor downregulation and postreceptor desensitization of signaling pathways; functional forms of resistance are generally reversible.
CLINICAL EVALUATION OF ENDOCRINE DISORDERS
Because most glands are relatively inaccessible, the physical examination usually focuses on the manifestations of hormone excess or deficiency as well as direct examination of palpable glands, such as the thyroid and gonads. For these reasons, it is important to evaluate patients in the context of their presenting symptoms, review of systems, family and social history, and exposure to medications that may affect the endocrine system. Astute clinical skills are required to detect subtle symptoms and signs suggestive of underlying endocrine disease. For example, a patient with Cushing’s syndrome may manifest specific findings, such as central fat redistribution, striae, and proximal muscle weakness, in addition to features seen commonly in the general population, such as obesity, plethora, hypertension, and glucose intolerance. Similarly, the insidious onset of hypothyroidism—with mental slowing, fatigue, dry skin, and other features—can be difficult to distinguish from similar, nonspecific findings in the general population. Clinical judgment that is based on knowledge of disease prevalence and pathophysiology is required to decide when to embark on more extensive evaluation of these disorders. Laboratory testing plays an essential role in endocrinology by allowing quantitative assessment of hormone levels and dynamics. Radiologic imaging tests such as computed tomography (CT) scan, magnetic resonance imaging (MRI), thyroid scan, and ultrasound are also used for the diagnosis of endocrine disorders. However, these tests generally are employed only after a hormonal abnormality has been established by biochemical testing.
HORMONE MEASUREMENTS AND ENDOCRINE TESTING
Immunoassays are the most important diagnostic tool in endocrinology, as they allow sensitive, specific, and quantitative determination of steady-state and dynamic changes in hormone concentrations. Immunoassays use antibodies to detect specific hormones. For many peptide hormones, these measurements are now configured to use two different antibodies to increase binding affinity and specificity. There are many variations of these assays; a common format involves using one antibody to capture the antigen (hormone) onto an immobilized surface and a second antibody, coupled to a chemiluminescent (immunochemiluminescent assay [ICMA]) or radioactive (immunoradiometric assay [IRMA]) signal, to detect the antigen. These assays are sensitive enough to detect plasma hormone concentrations in the picomolar to nanomolar range, and they can readily distinguish structurally related proteins, such as PTH from PTH-related peptide (PTHrP). A variety of other techniques are used to measure specific hormones, including mass spectroscopy, various forms of chromatography, and enzymatic methods; bioassays are now rarely used.
Most hormone measurements are based on plasma or serum samples. However, urinary hormone determinations remain useful for the evaluation of some conditions. Urinary collections over 24 h provide an integrated assessment of the production of a hormone or metabolite, many of which vary during the day. It is important to assure complete collections of 24-h urine samples; simultaneous measurement of creatinine provides an internal control for the adequacy of collection and can be used to normalize some hormone measurements. A 24-h urine free cortisol measurement largely reflects the amount of unbound cortisol, thus providing a reasonable index of biologically available hormone. Other commonly used urine determinations include 17-hydroxycorticosteroids, 17-ketosteroids, vanillylmandelic acid, metanephrine, catecholamines, 5-hydroxyindoleacetic acid, and calcium.
The value of quantitative hormone measurements lies in their correct interpretation in a clinical context. The normal range for most hormones is relatively broad, often varying by a factor of two- to tenfold. The normal ranges for many hormones are sex- and age-specific. Thus, using the correct normative database is an essential part of interpreting hormone tests. The pulsatile nature of hormones and factors that can affect their secretion, such as sleep, meals, and medications, must also be considered. Cortisol values increase fivefold between midnight and dawn; reproductive hormone levels vary dramatically during the female menstrual cycle.
For many endocrine systems, much information can be gained from basal hormone testing, particularly when different components of an endocrine axis are assessed simultaneously. For example, low testosterone and elevated LH levels suggest a primary gonadal problem, whereas a hypothalamic-pituitary disorder is likely if both LH and testosterone are low. Because TSH is a sensitive indicator of thyroid function, it is generally recommended as a first-line test for thyroid disorders. An elevated TSH level is almost always the result of primary hypothyroidism, whereas a low TSH is most often caused by thyrotoxicosis. These predictions can be confirmed by determining the free thyroxine level. In the less common circumstance when free thyroxine and TSH are both low, it is important to consider secondary hypopituitarism caused by hypothalamic-pituitary disease. Elevated calcium and PTH levels suggest hyperparathyroidism, whereas PTH is suppressed in hypercalcemia caused by malignancy or granulomatous diseases. A suppressed ACTH in the setting of hypercortisolemia, or increased urine free cortisol, is seen with hyperfunctioning adrenal adenomas.
It is not uncommon, however, for baseline hormone levels associated with pathologic endocrine conditions to overlap with the normal range. In this circumstance, dynamic testing is useful to separate the two groups further. There are a multitude of dynamic endocrine tests, but all are based on principles of feedback regulation, and most responses can be rationalized based on principles that govern the regulation of endocrine axes. Suppression tests are used in the setting of suspected endocrine hyperfunction. An example is the dexamethasone suppression test used to evaluate Cushing’s syndrome (Chaps. 403 and 406). Stimulation tests generally are used to assess endocrine hypofunction. The ACTH stimulation test, for example, is used to assess the adrenal gland response in patients with suspected adrenal insufficiency. Other stimulation tests use hypothalamic-releasing factors such as corticotropin-releasing hormone (CRH) and growth hormone–releasing hormone (GHRH) to evaluate pituitary hormone reserve (Chap. 403). Insulin-induced hypoglycemia also evokes pituitary ACTH and GH responses. Stimulation tests based on reduction or inhibition of endogenous hormones are now used infrequently. Examples include metyrapone inhibition of cortisol synthesis and clomiphene inhibition of estrogen feedback.
SCREENING AND ASSESSMENT OF COMMON ENDOCRINE DISORDERS
Many endocrine disorders are prevalent in the adult population (Table 399-2) and can be diagnosed and managed by general internists, family practitioners, or other primary health care providers. The high prevalence and clinical impact of certain endocrine diseases justifies vigilance for features of these disorders during routine physical examinations; laboratory screening is indicated in selected high-risk populations.
EXAMPLES OF PREVALENT ENDOCRINE AND METABOLIC DISORDERS IN THE ADULT |
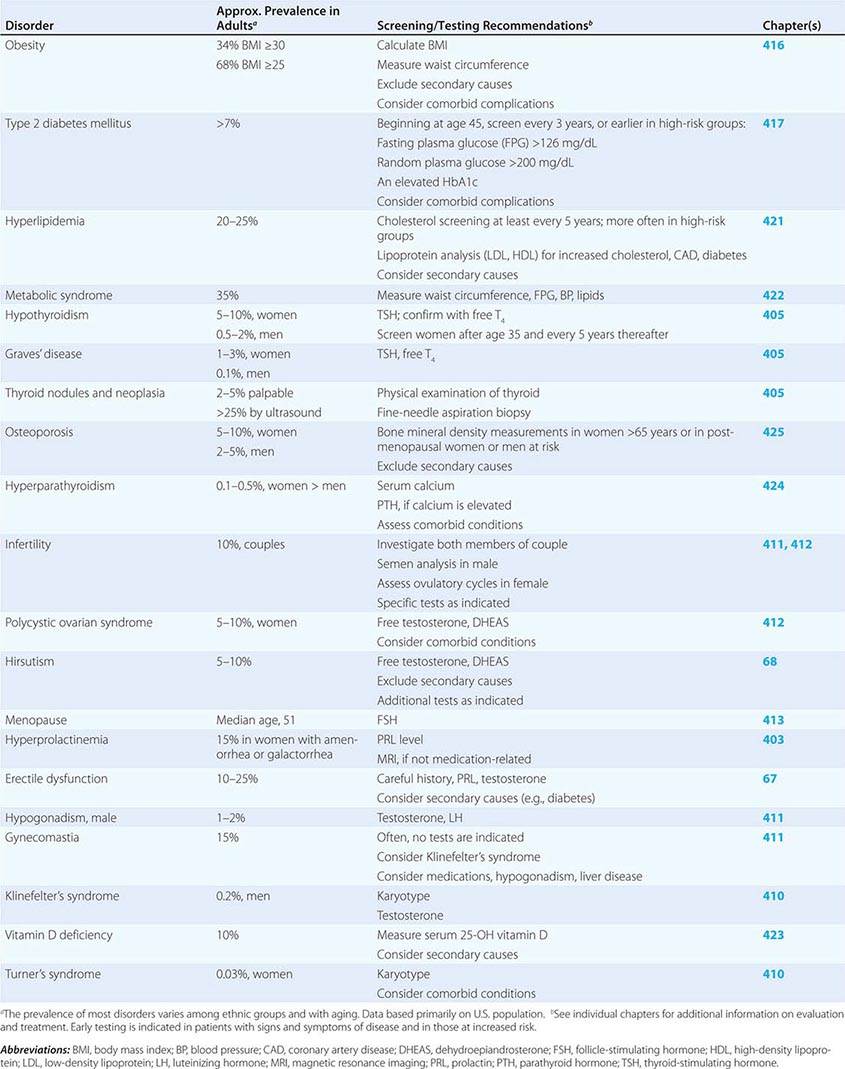
400e | Mechanisms of Hormone Action |
CLASSES OF HORMONES
Hormones can be divided into five major types: (1) amino acid derivatives such as dopamine, catecholamine, and thyroid hormone; (2) small neuropeptides such as gonadotropin-releasing hormone (GnRH), thyrotropin-releasing hormone (TRH), somatostatin, and vasopressin; (3) large proteins such as insulin, luteinizing hormone (LH), and parathyroid hormone (PTH); (4) steroid hormones such as cortisol and estrogen that are synthesized from cholesterol-based precursors; and (5) vitamin derivatives such as retinoids (vitamin A) and vitamin D. A variety of peptide growth factors, most of which act locally, share actions with hormones. As a rule, amino acid derivatives and peptide hormones interact with cell-surface membrane receptors. Steroids, thyroid hormones, vitamin D, and retinoids are lipid-soluble and interact with intracellular nuclear receptors, although many also interact with membrane receptors or intracellular signaling proteins as well.
HORMONE AND RECEPTOR FAMILIES
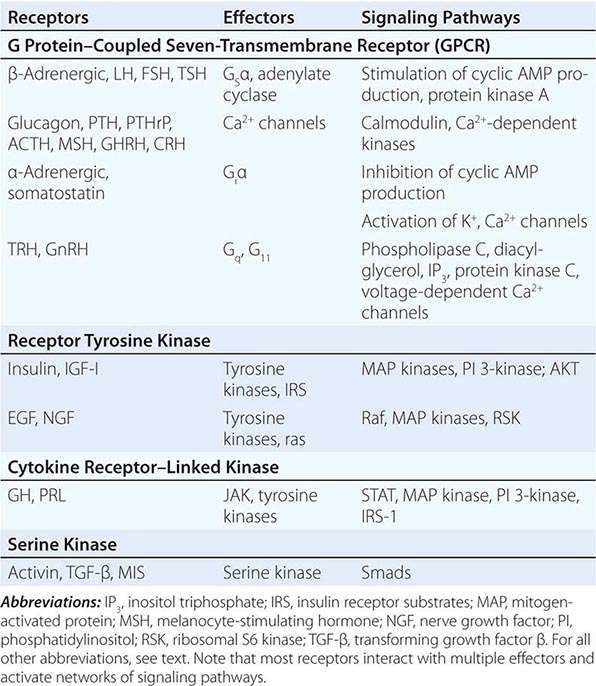
Hormones and receptors can be grouped into families, reflecting structural similarities and evolutionary origins (Table 400e-1). The evolution of these families generates diverse but highly selective pathways of hormone action. Recognition of these relationships has proven useful for extrapolating information gleaned from one hormone or receptor to other family members.
EXAMPLES OF MEMBRANE RECEPTOR FAMILIES AND SIGNALING PATHWAYS |
The glycoprotein hormone family, consisting of thyroid-stimulating hormone (TSH), follicle-stimulating hormone (FSH), LH, and human chorionic gonadotropin (hCG), illustrates many features of related hormones. The glycoprotein hormones are heterodimers that share the α subunit in common; the β subunits are distinct and confer specific biologic actions. The overall three-dimensional architecture of the β subunits is similar, reflecting the locations of conserved disulfide bonds that restrain protein conformation. The cloning of the β-subunit genes from multiple species suggests that this family arose from a common ancestral gene, probably by gene duplication and subsequent divergence to evolve new biologic functions.
As hormone families enlarge and diverge, their receptors must co-evolve to derive new biologic functions. Related G protein–coupled receptors (GPCRs), for example, have evolved for each of the glycoprotein hormones. These receptors are structurally similar, and each is coupled predominantly to the Gsα signaling pathway. However, there is minimal overlap of hormone binding. For example, TSH binds with high specificity to the TSH receptor but interacts minimally with the LH or FSH receptors. Nonetheless, there can be subtle physiologic consequences of hormone cross-reactivity with other receptors. Very high levels of hCG during pregnancy stimulate the TSH receptor and increase thyroid hormone levels, resulting in a compensatory decrease in TSH.
Insulin and insulin-like growth factor I (IGF-I) and IGF-II have structural similarities that are most apparent when precursor forms of the proteins are compared. In contrast to the high degree of specificity seen with the glycoprotein hormones, there is moderate cross-talk among the members of the insulin/IGF family. High concentrations of an IGF-II precursor produced by certain tumors (e.g., sarcomas) can cause hypoglycemia, partly because of binding to insulin and IGF-I receptors (Chap. 424). High concentrations of insulin also bind to the IGF-I receptor, perhaps accounting for some of the clinical manifestations seen in conditions with chronic hyperinsulinemia.
Another important example of receptor cross-talk is seen with PTH and parathyroid hormone–related peptide (PTHrP) (Chap. 424). PTH is produced by the parathyroid glands, whereas PTHrP is expressed at high levels during development and by a variety of tumors (Chap. 121). These hormones have amino acid sequence similarity, particularly in their amino-terminal regions. Both hormones bind to a single PTH receptor that is expressed in bone and kidney. Hypercalcemia and hypophosphatemia therefore may result from excessive production of either hormone, making it difficult to distinguish hyperparathyroidism from hypercalcemia of malignancy solely on the basis of serum chemistries. However, sensitive and specific assays for PTH and PTHrP now allow these disorders to be distinguished more readily.
Based on their specificities for DNA binding sites, the nuclear receptor family can be subdivided into type 1 receptors (glucocorticoid receptor, mineralocorticoid receptor, androgen receptor, estrogen receptor, progesterone receptor) that bind steroids and type 2 receptors (thyroid hormone receptor, vitamin D receptor, retinoic acid receptor, peroxisome proliferator activated receptor) that bind thyroid hormone, vitamin D, retinoic acid, or lipid derivatives. Certain functional domains in nuclear receptors, such as the zinc finger DNA-binding domains, are highly conserved. However, selective amino acid differences within this domain confer DNA sequence specificity. The hormone-binding domains are more variable, providing great diversity in the array of small molecules that bind to different nuclear receptors. With few exceptions, hormone binding is highly specific for a single type of nuclear receptor. One exception involves the glucocorticoid and mineralocorticoid receptors. Because the mineralocorticoid receptor also binds glucocorticoids with high affinity, an enzyme (11β-hydroxysteroid dehydrogenase) in renal tubular cells inactivates glucocorticoids, allowing selective responses to mineralocorticoids such as aldosterone. However, when very high glucocorticoid concentrations occur, as in Cushing’s syndrome, the glucocorticoid degradation pathway becomes saturated, allowing excessive cortisol levels to exert mineralocorticoid effects (sodium retention, potassium wasting). This phenomenon is particularly pronounced in ectopic adrenocorticotropic hormone (ACTH) syndromes (Chap. 406). Another example of relaxed nuclear receptor specificity involves the estrogen receptor, which can bind an array of compounds, some of which have little apparent structural similarity to the high-affinity ligand estradiol. This feature of the estrogen receptor makes it susceptible to activation by “environmental estrogens” such as resveratrol, octylphenol, and many other aromatic hydrocarbons. However, this lack of specificity provides an opportunity to synthesize a remarkable series of clinically useful antagonists (e.g., tamoxifen) and selective estrogen response modulators (SERMs) such as raloxifene. These compounds generate distinct conformations that alter receptor interactions with components of the transcription machinery (see below), thereby conferring their unique actions.
HORMONE SYNTHESIS AND PROCESSING
The synthesis of peptide hormones and their receptors occurs through a classic pathway of gene expression: transcription → mRNA → protein → posttranslational protein processing → intracellular sorting, followed by membrane integration or secretion (Chap. 82).
Many hormones are embedded within larger precursor polypeptides that are proteolytically processed to yield the biologically active hormone. Examples include proopiomelanocortin (POMC) → ACTH; proglucagon → glucagon; proinsulin → insulin; and pro-PTH → PTH, among others. In many cases, such as POMC and proglucagon, these precursors generate multiple biologically active peptides. It is provocative that hormone precursors are typically inactive, presumably adding an additional level of regulatory control. Prohormone conversion occurs not only for peptide hormones but also for certain steroids (testosterone → dihydrotestosterone) and thyroid hormone (T4 → T3).
Peptide precursor processing is intimately linked to intracellular sorting pathways that transport proteins to appropriate vesicles and enzymes, resulting in specific cleavage steps, followed by protein folding and translocation to secretory vesicles. Hormones destined for secretion are translocated across the endoplasmic reticulum under the guidance of an amino-terminal signal sequence that subsequently is cleaved. Cell-surface receptors are inserted into the membrane via short segments of hydrophobic amino acids that remain embedded within the lipid bilayer. During translocation through the Golgi and endoplasmic reticulum, hormones and receptors are subject to a variety of posttranslational modifications, such as glycosylation and phosphorylation, which can alter protein conformation, modify circulating half-life, and alter biologic activity.
Synthesis of most steroid hormones is based on modifications of the precursor, cholesterol. Multiple regulated enzymatic steps are required for the synthesis of testosterone (Chap. 411), estradiol (Chap. 412), cortisol (Chap. 406), and vitamin D (Chap. 423). This large number of synthetic steps predisposes to multiple genetic and acquired disorders of steroidogenesis.
Endocrine genes contain regulatory DNA elements similar to those found in many other genes, but their exquisite control by hormones reflects the presence of specific hormone response elements. For example, the TSH genes are repressed directly by thyroid hormones acting through the thyroid hormone receptor (TR), a member of the nuclear receptor family. Steroidogenic enzyme gene expression requires specific transcription factors, such as steroidogenic factor-1 (SF-1), acting in conjunction with signals transmitted by trophic hormones (e.g., ACTH or LH). For some hormones, substantial regulation occurs at the level of translational efficiency. Insulin biosynthesis, although it requires ongoing gene transcription, is regulated primarily at the translational and secretory levels in response to elevated levels of glucose or amino acids.
HORMONE SECRETION, TRANSPORT, AND DEGRADATION
The level of a hormone is determined by its rate of secretion and its circulating half-life. After protein processing, peptide hormones (e.g., GnRH, insulin, growth hormone [GH]) are stored in secretory granules. As these granules mature, they are poised beneath the plasma membrane for imminent release into the circulation. In most instances, the stimulus for hormone secretion is a releasing factor or neural signal that induces rapid changes in intracellular calcium concentrations, leading to secretory granule fusion with the plasma membrane and release of its contents into the extracellular environment and bloodstream. Steroid hormones, in contrast, diffuse into the circulation as they are synthesized. Thus, their secretory rates are closely aligned with rates of synthesis. For example, ACTH and LH induce steroidogenesis by stimulating the activity of the steroidogenic acute regulatory (StAR) protein (transports cholesterol into the mitochondrion) along with other rate-limiting steps (e.g., cholesterol side-chain cleavage enzyme, CYP11A1) in the steroidogenic pathway.
Hormone transport and degradation dictate the rapidity with which a hormonal signal decays. Some hormone signals are evanescent (e.g., somatostatin), whereas others are longer-lived (e.g., TSH). Because somatostatin exerts effects in virtually every tissue, a short half-life allows its concentrations and actions to be controlled locally. Structural modifications that impair somatostatin degradation have been useful for generating long-acting therapeutic analogues such as octreotide (Chap. 403). In contrast, the actions of TSH are highly specific for the thyroid gland. Its prolonged half-life accounts for relatively constant serum levels even though TSH is secreted in discrete pulses.
An understanding of circulating hormone half-life is important for achieving physiologic hormone replacement, as the frequency of dosing and the time required to reach steady state are intimately linked to rates of hormone decay. T4, for example, has a circulating half-life of 7 days. Consequently, >1 month is required to reach a new steady state, and single daily doses are sufficient to achieve constant hormone levels. T3, in contrast, has a half-life of 1 day. Its administration is associated with more dynamic serum levels, and it must be administered two to three times per day. Similarly, synthetic glucocorticoids vary widely in their half-lives; those with longer half-lives (e.g., dexamethasone) are associated with greater suppression of the hypothalamic-pituitary-adrenal (HPA) axis. Most protein hormones (e.g., ACTH, GH, prolactin [PRL], PTH, LH) have relatively short half-lives (<20 min), leading to sharp peaks of secretion and decay. The only accurate way to profile the pulse frequency and amplitude of these hormones is to measure levels in frequently sampled blood (every 10 min or less) over long durations (8–24 h). Because this is not practical in a clinical setting, an alternative strategy is to pool three to four samples drawn at about 30-min intervals, or interpret the results in the context of a relatively wide normal range. Rapid hormone decay is useful in certain clinical settings. For example, the short half-life of PTH allows the use of intraoperative PTH determinations to confirm successful removal of an adenoma. This is particularly valuable diagnostically when there is a possibility of multicentric disease or parathyroid hyperplasia, as occurs with multiple endocrine neoplasia (MEN) or renal insufficiency.
Many hormones circulate in association with serum-binding proteins. Examples include (1) T4 and T3 binding to thyroxine-binding globulin (TBG), albumin, and thyroxine-binding prealbumin (TBPA); (2) cortisol binding to cortisol-binding globulin (CBG); (3) androgen and estrogen binding to sex hormone–binding globulin (SHBG); (4) IGF-I and -II binding to multiple IGF-binding proteins (IGFBPs); (5) GH interactions with GH-binding protein (GHBP), a circulating fragment of the GH receptor extracellular domain; and (6) activin binding to follistatin. These interactions provide a hormonal reservoir, prevent otherwise rapid degradation of unbound hormones, restrict hormone access to certain sites (e.g., IGFBPs), and modulate the unbound, or “free,” hormone concentrations. Although a variety of binding protein abnormalities have been identified, most have little clinical consequence aside from creating diagnostic problems. For example, TBG deficiency can reduce total thyroid hormone levels greatly but the free concentrations of T4 and T3 remain normal. Liver disease and certain medications can also influence binding protein levels (e.g., estrogen increases TBG) or cause displacement of hormones from binding proteins (e.g., salsalate displaces T4 from TBG). In general, only unbound hormone is available to interact with receptors and thus elicit a biologic response. Short-term perturbations in binding proteins change the free hormone concentration, which in turn induces compensatory adaptations through feedback loops. SHBG changes in women are an exception to this self-correcting mechanism. When SHBG decreases because of insulin resistance or androgen excess, the unbound testosterone concentration is increased, potentially leading to hirsutism (Chap. 68). The increased unbound testosterone level does not result in an adequate compensatory feedback correction because estrogen, not testosterone, is the primary regulator of the reproductive axis.
An additional exception to the unbound hormone hypothesis involves megalin, a member of the low-density lipoprotein (LDL) receptor family that serves as an endocytotic receptor for carrier-bound vitamins A and D and SHBG-bound androgens and estrogens. After internalization, the carrier proteins are degraded in lysosomes and release their bound ligands within the cells. Membrane transporters have also been identified for thyroid hormones.
Hormone degradation can be an important mechanism for regulating concentrations locally. As noted above, 11β-hydroxysteroid dehydrogenase inactivates glucocorticoids in renal tubular cells, preventing actions through the mineralocorticoid receptor. Thyroid hormone deiodinases convert T4 to T3 and can inactivate T3. During development, degradation of retinoic acid by Cyp26b1 prevents primordial germ cells in the male from entering meiosis, as occurs in the female ovary.
HORMONE ACTION THROUGH RECEPTORS
Receptors for hormones are divided into two major classes: membrane and nuclear. Membrane receptors primarily bind peptide hormones and catecholamines. Nuclear receptors bind small molecules that can diffuse across the cell membrane, such as steroids and vitamin D. Certain general principles apply to hormone-receptor interactions regardless of the class of receptor. Hormones bind to receptors with specificity and an affinity that generally coincides with the dynamic range of circulating hormone concentrations. Low concentrations of free hormone (usually 10–12 to 10–9 M) rapidly associate and dissociate from receptors in a bimolecular reaction such that the occupancy of the receptor at any given moment is a function of hormone concentration and the receptor’s affinity for the hormone. Receptor numbers vary greatly in different target tissues, providing one of the major determinants of specific tissue responses to circulating hormones. For example, ACTH receptors are located almost exclusively in the adrenal cortex, and FSH receptors are found predominantly in the gonads. In contrast, insulin and TRs are widely distributed, reflecting the need for metabolic responses in all tissues.
MEMBRANE RECEPTORS
Membrane receptors for hormones can be divided into several major groups: (1) seven transmembrane GPCRs, (2) tyrosine kinase receptors, (3) cytokine receptors, and (4) serine kinase receptors (Fig. 400e-1). The seven transmembrane GPCR family binds a remarkable array of hormones, including large proteins (e.g., LH, PTH), small peptides (e.g., TRH, somatostatin), catecholamines (epinephrine, dopamine), and even minerals (e.g., calcium). The extracellular domains of GPCRs vary widely in size and are the major binding site for large hormones. The transmembrane-spanning regions are composed of hydrophobic α-helical domains that traverse the lipid bilayer. Like some channels, these domains are thought to circularize and form a hydrophobic pocket into which certain small ligands fit. Hormone binding induces conformational changes in these domains, transducing structural changes to the intracellular domain, which is a docking site for G proteins.
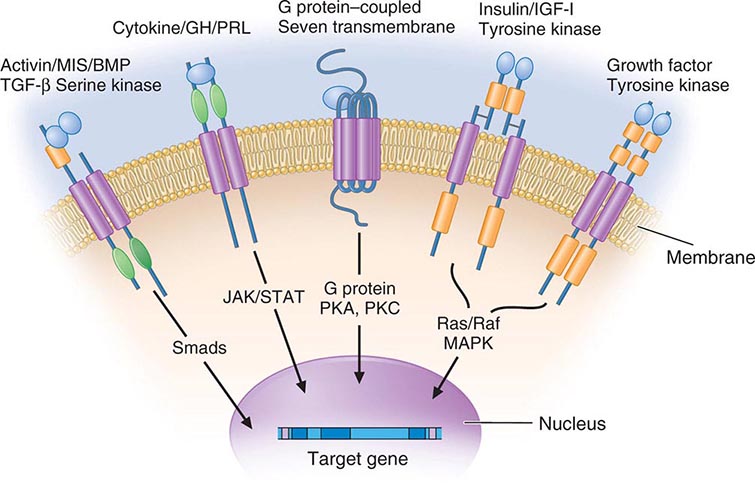
FIGURE 400e-1 Membrane receptor signaling. MAPK, mitogen-activated protein kinase; PKA, C, protein kinase A, C; TGF, transforming growth factor. For other abbreviations, see text.
The large family of G proteins, so named because they bind guanine nucleotides (guanosine triphosphate [GTP], guanosine diphosphate [GDP]), provides great diversity for coupling receptors to different signaling pathways. G proteins form a heterotrimeric complex that is composed of various α and βγ subunits. The α subunit contains the guanine nucleotide–binding site and hydrolyzes GTP → GDP. The βγ subunits are tightly associated and modulate the activity of the α subunit as well as mediating their own effector signaling pathways. G protein activity is regulated by a cycle that involves GTP hydrolysis and dynamic interactions between the α and αβ subunits. Hormone binding to the receptor induces GDP dissociation, allowing Gα to bind GTP and dissociate from the αβ complex. Under these conditions, the Gα subunit is activated and mediates signal transduction through various enzymes, such as adenylate cyclase and phospholipase C. GTP hydrolysis to GDP allows reassociation with the βγ subunits and restores the inactive state. As described below, a variety of endocrinopathies result from G protein mutations or from mutations in receptors that modify their interactions with G proteins. G proteins interact with other cellular proteins, including kinases, channels, G protein–coupled receptor kinases (GRKs), and arrestins, that mediate signaling as well as receptor desensitization and recycling.
The tyrosine kinase receptors transduce signals for insulin and a variety of growth factors, such as IGF-I, epidermal growth factor (EGF), nerve growth factor, platelet-derived growth factor, and fibroblast growth factor. The cysteine-rich extracellular ligand-binding domains contain growth factor binding sites. After ligand binding, this class of receptors undergoes autophosphorylation, inducing interactions with intracellular adaptor proteins such as Shc and insulin receptor substrates (IRS). In the case of the insulin receptor, multiple kinases are activated, including the Raf-Ras-MAPK and the Akt/protein kinase B pathways. The tyrosine kinase receptors play a prominent role in cell growth and differentiation as well as in intermediary metabolism.
The GH and PRL receptors belong to the cytokine receptor family. Analogous to the tyrosine kinase receptors, ligand binding induces receptor interaction with intracellular kinases—the Janus kinases (JAKs), which phosphorylate members of the signal transduction and activators of transcription (STAT) family—as well as with other signaling pathways (Ras, PI3-K, MAPK). The activated STAT proteins translocate to the nucleus and stimulate expression of target genes.
The serine kinase receptors mediate the actions of activins, transforming growth factor β, müllerian-inhibiting substance (MIS, also known as anti-müllerian hormone, AMH), and bone morphogenic proteins (BMPs). This family of receptors (consisting of type I and II subunits) signals through proteins termed smads (fusion of terms for Caenorhabditis elegans sma + mammalian mad). Like the STAT proteins, the smads serve a dual role of transducing the receptor signal and acting as transcription factors. The pleomorphic actions of these growth factors dictate that they act primarily in a local (paracrine or autocrine) manner. Binding proteins such as follistatin (which binds activin and other members of this family) function to inactivate the growth factors and restrict their distribution.
NUCLEAR RECEPTORS
The family of nuclear receptors has grown to nearly 100 members, many of which are still classified as orphan receptors because their ligands, if they exist, have not been identified (Fig. 400e-2). Otherwise, most nuclear receptors are classified on the basis of their ligands. Although all nuclear receptors ultimately act to increase or decrease gene transcription, some (e.g., glucocorticoid receptor) reside primarily in the cytoplasm, whereas others (e.g., TR) are located in the nucleus. After ligand binding, the cytoplasmically localized receptors translocate to the nucleus. There is growing evidence that certain nuclear receptors (e.g., glucocorticoid, estrogen) can also act at the membrane or in the cytoplasm to activate or repress signal transduction pathways, providing a mechanism for cross-talk between membrane and nuclear receptors.
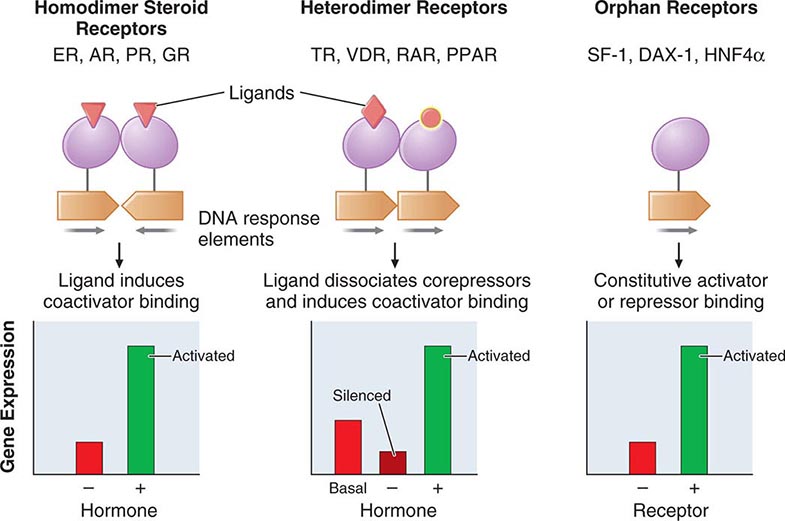
FIGURE 400e-2 Nuclear receptor signaling. AR, androgen receptor; DAX, dosage-sensitive sex-reversal, adrenal hypoplasia congenita, X-chromosome; ER, estrogen receptor; GR, glucocorticoid receptor; HNF4α, hepatic nuclear factor 4α; PPAR, peroxisome proliferator activated receptor; PR, progesterone receptor; RAR, retinoic acid receptor; SF-1, steroidogenic factor-1; TR, thyroid hormone receptor; VDR, vitamin D receptor.
The structures of nuclear receptors have been studied extensively, including by x-ray crystallography. The DNA binding domain, consisting of two zinc fingers, contacts specific DNA recognition sequences in target genes. Most nuclear receptors bind to DNA as dimers. Consequently, each monomer recognizes an individual DNA motif, referred to as a “half-site.” The steroid receptors, including the glucocorticoid, estrogen, progesterone, and androgen receptors, bind to DNA as homodimers. Consistent with this twofold symmetry, their DNA recognition half-sites are palindromic. The thyroid, retinoid, peroxisome proliferator activated, and vitamin D receptors bind to DNA preferentially as heterodimers in combination with retinoid × receptors (RXRs). Their DNA half-sites are typically arranged as direct repeats.
The carboxy-terminal hormone-binding domain mediates transcriptional control. For type II receptors such as TR and retinoic acid receptor (RAR), co-repressor proteins bind to the receptor in the absence of ligand and silence gene transcription. Hormone binding induces conformational changes, triggering the release of co-repressors and inducing the recruitment of coactivators that stimulate transcription. Thus, these receptors are capable of mediating dramatic changes in the level of gene activity. Certain disease states are associated with defective regulation of these events. For example, mutations in the TR prevent co-repressor dissociation, resulting in an autosomal dominant form of hormone resistance (Chap. 405). In promyelocytic leukemia, fusion of RARα to other nuclear proteins causes aberrant gene silencing that prevents normal cellular differentiation. Treatment with retinoic acid reverses this repression and allows cellular differentiation and apoptosis to occur. Most type 1 steroid receptors interact weakly with co-repressors, but ligand binding still induces interactions with an array of coactivators. X-ray crystallography shows that various SERMs induce distinct estrogen receptor conformations. The tissue-specific responses caused by these agents in breast, bone, and uterus appear to reflect distinct interactions with coactivators. The receptor-coactivator complex stimulates gene transcription by several pathways, including (1) recruitment of enzymes (histone acetyl transferases) that modify chromatin structure, (2) interactions with additional transcription factors on the target gene, and (3) direct interactions with components of the general transcription apparatus to enhance the rate of RNA polymerase II–mediated transcription. Studies of nuclear receptor-mediated transcription show that these are dynamic events that involve relatively rapid (e.g., 30–60 min) cycling of transcription complexes on any specific target gene.
FUNCTIONS OF HORMONES
The functions of individual hormones are described in detail in subsequent chapters. Nevertheless, it is useful to illustrate how most biologic responses require integration of several different hormone pathways. The physiologic functions of hormones can be divided into three general areas: (1) growth and differentiation, (2) maintenance of homeostasis, and (3) reproduction.
GROWTH
Multiple hormones and nutritional factors mediate the complex phenomenon of growth (Chap. 401e). Short stature may be caused by GH deficiency, hypothyroidism, Cushing’s syndrome, precocious puberty, malnutrition, chronic illness, or genetic abnormalities that affect the epiphyseal growth plates (e.g., FGFR3 and SHOX mutations). Many factors (GH, IGF-I, thyroid hormones) stimulate growth, whereas others (sex steroids) lead to epiphyseal closure. Understanding these hormonal interactions is important in the diagnosis and management of growth disorders. For example, delaying exposure to high levels of sex steroids may enhance the efficacy of GH treatment.
MAINTENANCE OF HOMEOSTASIS
Although virtually all hormones affect homeostasis, the most important among them are the following:
1. Thyroid hormone—controls about 25% of basal metabolism in most tissues
2. Cortisol—exerts a permissive action for many hormones in addition to its own direct effects
3. PTH—regulates calcium and phosphorus levels
4. Vasopressin—regulates serum osmolality by controlling renal free-water clearance
5. Mineralocorticoids—control vascular volume and serum electrolyte (Na+, K+) concentrations
6. Insulin—maintains euglycemia in the fed and fasted states
The defense against hypoglycemia is an impressive example of integrated hormone action (Chap. 420). In response to the fasting state and falling blood glucose, insulin secretion is suppressed, resulting in decreased glucose uptake and enhanced glycogenolysis, lipolysis, proteolysis, and gluconeogenesis to mobilize fuel sources. If hypoglycemia develops (usually from insulin administration or sulfonylureas), an orchestrated counterregulatory response occurs—glucagon and epinephrine rapidly stimulate glycogenolysis and gluconeogenesis, whereas GH and cortisol act over several hours to raise glucose levels and antagonize insulin action.
Although free-water clearance is controlled primarily by vasopressin, cortisol and thyroid hormone are also important for facilitating renal tubular responses to vasopressin (Chap. 404). PTH and vitamin D function in an interdependent manner to control calcium metabolism (Chap. 423). PTH stimulates renal synthesis of 1,25-dihydroxyvitamin D, which increases calcium absorption in the gastrointestinal tract and enhances PTH action in bone. Increased calcium, along with vitamin D, feeds back to suppress PTH, thus maintaining calcium balance.
Depending on the severity of a specific stress and whether it is acute or chronic, multiple endocrine and cytokine pathways are activated to mount an appropriate physiologic response. In severe acute stress such as trauma or shock, the sympathetic nervous system is activated and catecholamines are released, leading to increased cardiac output and a primed musculoskeletal system. Catecholamines also increase mean blood pressure and stimulate glucose production. Multiple stress-induced pathways converge on the hypothalamus, stimulating several hormones, including vasopressin and corticotropin-releasing hormone (CRH). These hormones, in addition to cytokines (tumor necrosis factor α, interleukin [IL] 2, IL-6) increase ACTH and GH production. ACTH stimulates the adrenal gland, increasing cortisol, which in turn helps sustain blood pressure and dampen the inflammatory response. Increased vasopressin acts to conserve free water.
REPRODUCTION
The stages of reproduction include (1) sex determination during fetal development (Chap. 410); (2) sexual maturation during puberty (Chaps. 411 and 412); (3) conception, pregnancy, lactation, and child rearing (Chap. 412); and (4) cessation of reproductive capability at menopause (Chap. 413). Each of these stages involves an orchestrated interplay of multiple hormones, a phenomenon well illustrated by the dynamic hormonal changes that occur during each 28-day menstrual cycle. In the early follicular phase, pulsatile secretion of LH and FSH stimulates the progressive maturation of the ovarian follicle. This results in gradually increasing estrogen and progesterone levels, leading to enhanced pituitary sensitivity to GnRH, which, when combined with accelerated GnRH secretion, triggers the LH surge and rupture of the mature follicle. Inhibin, a protein produced by the granulosa cells, enhances follicular growth and feeds back to the pituitary to selectively suppress FSH without affecting LH. Growth factors such as EGF and IGF-I modulate follicular responsiveness to gonadotropins. Vascular endothelial growth factor and prostaglandins play a role in follicle vascularization and rupture.
During pregnancy, the increased production of prolactin, in combination with placentally derived steroids (e.g., estrogen and progesterone), prepares the breast for lactation. Estrogens induce the production of progesterone receptors, allowing for increased responsiveness to progesterone. In addition to these and other hormones involved in lactation, the nervous system and oxytocin mediate the suckling response and milk release.
HORMONAL FEEDBACK REGULATORY SYSTEMS
Feedback control, both negative and positive, is a fundamental feature of endocrine systems. Each of the major hypothalamic-pituitary-hormone axes is governed by negative feedback, a process that maintains hormone levels within a relatively narrow range (Chap. 401e). Examples of hypothalamic-pituitary negative feedback include (1) thyroid hormones on the TRH-TSH axis, (2) cortisol on the CRH-ACTH axis, (3) gonadal steroids on the GnRH-LH/FSH axis, and (4) IGF-I on the growth hormone–releasing hormone (GHRH)-GH axis (Fig. 400e-3). These regulatory loops include both positive (e.g., TRH, TSH) and negative (e.g., T4, T3) components, allowing for exquisite control of hormone levels. As an example, a small reduction of thyroid hormone triggers a rapid increase of TRH and TSH secretion, resulting in thyroid gland stimulation and increased thyroid hormone production. When thyroid hormone reaches a normal level, it feeds back to suppress TRH and TSH, and a new steady state is attained. Feedback regulation also occurs for endocrine systems that do not involve the pituitary gland, such as calcium feedback on PTH, glucose inhibition of insulin secretion, and leptin feedback on the hypothalamus. An understanding of feedback regulation provides important insights into endocrine testing paradigms (see below).
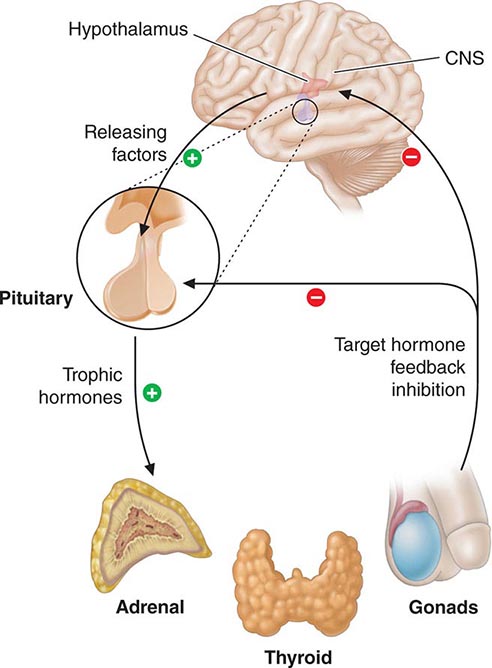
FIGURE 400e-3 Feedback regulation of endocrine axes. CNS, central nervous system.
Positive feedback control also occurs but is not well understood. The primary example is estrogen-mediated stimulation of the midcycle LH surge. Although chronic low levels of estrogen are inhibitory, gradually rising estrogen levels stimulate LH secretion. This effect, which is illustrative of an endocrine rhythm (see below), involves activation of the hypothalamic GnRH pulse generator. In addition, estrogen-primed gonadotropes are extraordinarily sensitive to GnRH, leading to amplification of LH release.
PARACRINE AND AUTOCRINE CONTROL
The previously mentioned examples of feedback control involve classic endocrine pathways in which hormones are released by one gland and act on a distant target gland. However, local regulatory systems, often involving growth factors, are increasingly recognized. Paracrine regulation refers to factors released by one cell that act on an adjacent cell in the same tissue. For example, somatostatin secretion by pancreatic islet δ cells inhibits insulin secretion from nearby β cells. Autocrine regulation describes the action of a factor on the same cell from which it is produced. IGF-I acts on many cells that produce it, including chondrocytes, breast epithelium, and gonadal cells. Unlike endocrine actions, paracrine and autocrine control are difficult to document because local growth factor concentrations cannot be measured readily.
Anatomic relationships of glandular systems also greatly influence hormonal exposure: the physical organization of islet cells enhances their intercellular communication; the portal vasculature of the hypothalamic-pituitary system exposes the pituitary to high concentrations of hypothalamic releasing factors; testicular seminiferous tubules gain exposure to high testosterone levels produced by the interdigitated Leydig cells; the pancreas receives nutrient information and local exposure to peptide hormones (incretins) from the gastrointestinal tract; and the liver is the proximal target of insulin action because of portal drainage from the pancreas.
HORMONAL RHYTHMS
The feedback regulatory systems described above are superimposed on hormonal rhythms that are used for adaptation to the environment. Seasonal changes, the daily occurrence of the light-dark cycle, sleep, meals, and stress are examples of the many environmental events that affect hormonal rhythms. The menstrual cycle is repeated on average every 28 days, reflecting the time required to follicular maturation and ovulation (Chap. 412). Essentially all pituitary hormone rhythms are entrained to sleep and to the circadian cycle, generating reproducible patterns that are repeated approximately every 24 h. The HPA axis, for example, exhibits characteristic peaks of ACTH and cortisol production in the early morning, with a nadir during the night. Recognition of these rhythms is important for endocrine testing and treatment. Patients with Cushing’s syndrome characteristically exhibit increased midnight cortisol levels compared with normal individuals (Chap. 406). In contrast, morning cortisol levels are similar in these groups, as cortisol is normally high at this time of day in normal individuals. The HPA axis is more susceptible to suppression by glucocorticoids administered at night as they blunt the early-morning rise of ACTH. Understanding these rhythms allows glucocorticoid replacement that mimics diurnal production by administering larger doses in the morning than in the afternoon. Disrupted sleep rhythms can alter hormonal regulation. For example, sleep deprivation causes mild insulin resistance, food craving, and hypertension, which are reversible, at least in the short term. Emerging evidence indicates that circadian clock pathways not only regulate sleep-wake cycles but also play important roles in virtually every cell type. For example, tissue-specific deletion of clock genes alters rhythms and levels of gene expression, as well as metabolic responses in liver, adipose, and other tissues.
Other endocrine rhythms occur on a more rapid time scale. Many peptide hormones are secreted in discrete bursts every few hours. LH and FSH secretion are exquisitely sensitive to GnRH pulse frequency. Intermittent pulses of GnRH are required to maintain pituitary sensitivity, whereas continuous exposure to GnRH causes pituitary gonadotrope desensitization. This feature of the hypothalamic-pituitary-gonadotrope axis forms the basis for using long-acting GnRH agonists to treat central precocious puberty or to decrease testosterone levels in the management of prostate cancer. It is important to be aware of the pulsatile nature of hormone secretion and the rhythmic patterns of hormone production in relating serum hormone measurements to normal values. For some hormones, integrated markers have been developed to circumvent hormonal fluctuations. Examples include 24-h urine collections for cortisol, IGF-I as a biologic marker of GH action, and HbA1c as an index of long-term (weeks to months) blood glucose control.
Often, one must interpret endocrine data only in the context of other hormones. For example, PTH levels typically are assessed in combination with serum calcium concentrations. A high serum calcium level in association with elevated PTH is suggestive of hyperparathyroidism, whereas a suppressed PTH in this situation is more likely to be caused by hypercalcemia of malignancy or other causes of hypercalcemia. Similarly, TSH should be elevated when T4 and T3 concentrations are low, reflecting reduced feedback inhibition. When this is not the case, it is important to consider secondary hypothyroidism, which is caused by a defect at the level of the pituitary.
401e | Anterior Pituitary: Physiology of Pituitary Hormones |
The anterior pituitary often is referred to as the “master gland” because, together with the hypothalamus, it orchestrates the complex regulatory functions of many other endocrine glands. The anterior pituitary gland produces six major hormones: (1) prolactin (PRL), (2) growth hormone (GH), (3) adrenocorticotropic hormone (ACTH), (4) luteinizing hormone (LH), (5) follicle-stimulating hormone (FSH), and (6) thyroid-stimulating hormone (TSH) (Table 401e-1). Pituitary hormones are secreted in a pulsatile manner, reflecting stimulation by an array of specific hypothalamic releasing factors. Each of these pituitary hormones elicits specific responses in peripheral target tissues. The hormonal products of those peripheral glands, in turn, exert feedback control at the level of the hypothalamus and pituitary to modulate pituitary function (Fig. 401e-1). Pituitary tumors cause characteristic hormone excess syndromes. Hormone deficiency may be inherited or acquired. Fortunately, there are efficacious treatments for many pituitary hormone excess and deficiency syndromes. Nonetheless, these diagnoses are often elusive; this emphasizes the importance of recognizing subtle clinical manifestations and performing the correct laboratory diagnostic tests. For discussion of disorders of the posterior pituitary, or neurohypophysis, see Chap. 404.
ANTERIOR PITUITARY HORMONE EXPRESSION AND REGULATION |
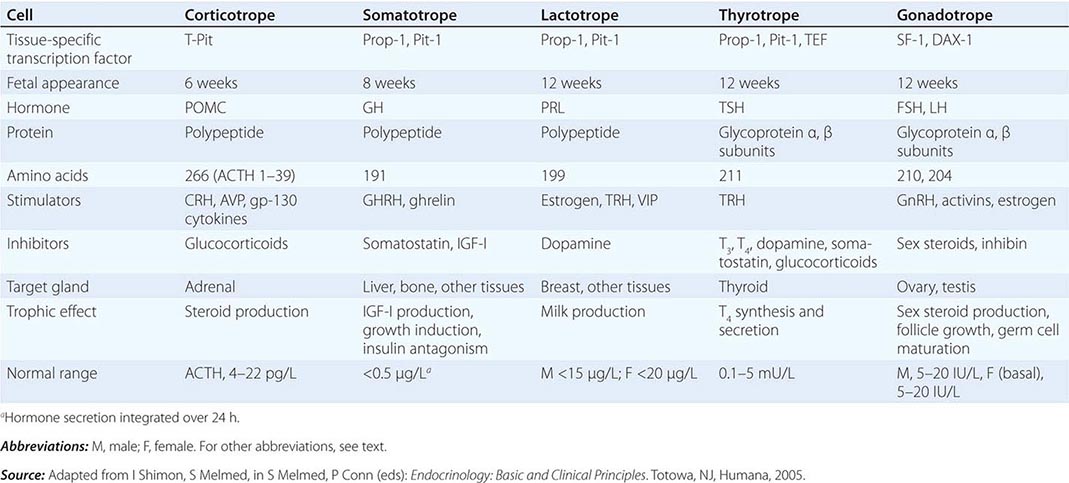
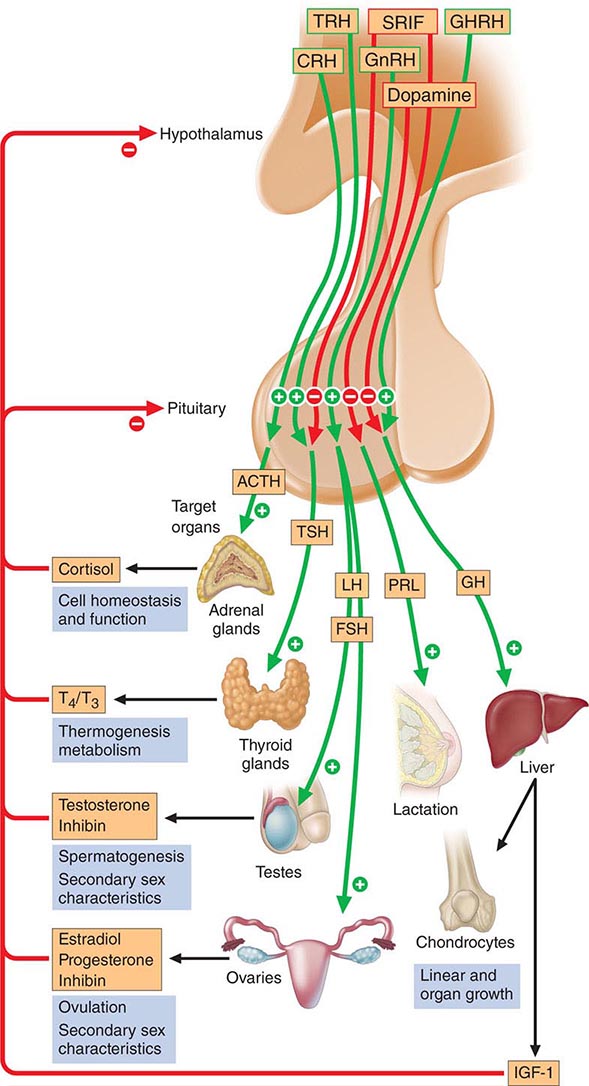
FIGURE 401e-1 Diagram of pituitary axes. Hypothalamic hormones regulate anterior pituitary trophic hormones that in turn determine target gland secretion. Peripheral hormones feed back to regulate hypothalamic and pituitary hormones. For abbreviations, see text.
ANATOMY AND DEVELOPMENT
ANATOMY
The pituitary gland weighs ~600 mg and is located within the sella turcica ventral to the diaphragma sella; it consists of anatomically and functionally distinct anterior and posterior lobes. The bony sella is contiguous to vascular and neurologic structures, including the cavernous sinuses, cranial nerves, and optic chiasm. Thus, expanding intrasellar pathologic processes may have significant central mass effects in addition to their endocrinologic impact.
Hypothalamic neural cells synthesize specific releasing and inhibiting hormones that are secreted directly into the portal vessels of the pituitary stalk. Blood supply of the pituitary gland comes from the superior and inferior hypophyseal arteries (Fig. 401e-2). The hypothalamic-pituitary portal plexus provides the major blood source for the anterior pituitary, allowing reliable transmission of hypothalamic peptide pulses without significant systemic dilution; consequently, pituitary cells are exposed to releasing or inhibiting factors and in turn release their hormones as discrete pulses into the systemic circulation (Fig. 401e-3).
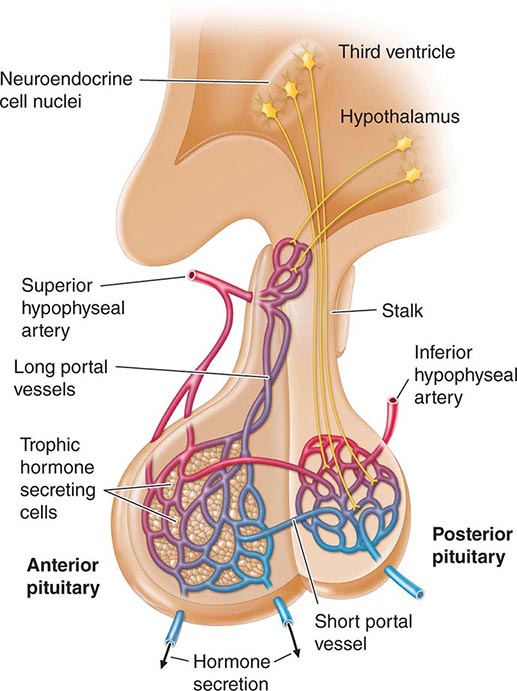
FIGURE 401e-2 Diagram of hypothalamic-pituitary vasculature. The hypothalamic nuclei produce hormones that traverse the portal system and impinge on anterior pituitary cells to regulate pituitary hormone secretion. Posterior pituitary hormones are derived from direct neural extensions.
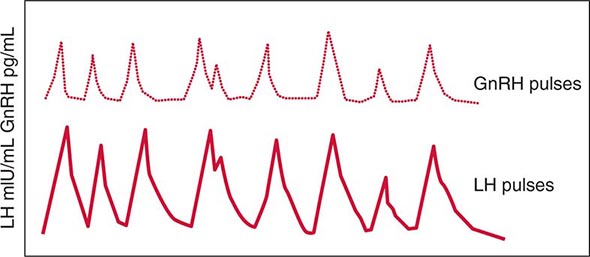
FIGURE 401e-3 Hypothalamic gonadotropin-releasing hormone (GnRH) pulses induce secretory pulses of luteinizing hormone (LH).
The posterior pituitary is supplied by the inferior hypophyseal arteries. In contrast to the anterior pituitary, the posterior lobe is directly innervated by hypothalamic neurons (supraopticohypophyseal and tuberohypophyseal nerve tracts) via the pituitary stalk (Chap. 404). Thus, posterior pituitary production of vasopressin (antidiuretic hormone [ADH]) and oxytocin is particularly sensitive to neuronal damage by lesions that affect the pituitary stalk or hypothalamus.
PITUITARY DEVELOPMENT
The embryonic differentiation and maturation of anterior pituitary cells have been elucidated in considerable detail. Pituitary development from Rathke’s pouch involves a complex interplay of lineage-specific transcription factors expressed in pluripotent precursor cells and gradients of locally produced growth factors (Table 401e-1). The transcription factor Prop-1 induces pituitary development of Pit-1-specific lineages as well as gonadotropes. The transcription factor Pit-1 determines cell-specific expression of GH, PRL, and TSH in somatotropes, lactotropes, and thyrotropes. Expression of high levels of estrogen receptors in cells that contain Pit-1 favors PRL expression, whereas thyrotrope embryonic factor (TEF) induces TSH expression. Pit-1 binds to GH, PRL, and TSH gene regulatory elements as well as to recognition sites on its own promoter, providing a mechanism for maintaining specific pituitary hormone phenotypic stability. Gonadotrope cell development is further defined by the cell-specific expression of the nuclear receptors steroidogenic factor (SF-1) and d osage-sensitive sex reversal, a drenal hypoplasia critical region, on chromosome X, gene 1 (DAX-1). Development of corticotrope cells, which express the proopiomelanocortin (POMC) gene, requires the T-Pit transcription factor. Abnormalities of pituitary development caused by mutations of Pit-1, Prop-1, SF-1, DAX-1, and T-Pit result in a rare, selective or combined pituitary hormone deficit syndromes.
ANTERIOR PITUITARY HORMONES
Each anterior pituitary hormone is under unique control, and each exhibits highly specific normal and dysregulated secretory characteristics.
PROLACTIN
Synthesis PRL consists of 198 amino acids and has a molecular mass of 21,500 kDa; it is weakly homologous to GH and human placental lactogen (hPL), reflecting the duplication and divergence of a common GH-PRL-hPL precursor gene. PRL is synthesized in lactotropes, which constitute about 20% of anterior pituitary cells. Lactotropes and somatotropes are derived from a common precursor cell that may give rise to a tumor that secretes both PRL and GH. Marked lactotrope cell hyperplasia develops during pregnancy and the first few months of lactation. These transient functional changes in the lactotrope population are induced by estrogen.
Secretion Normal adult serum PRL levels are about 10–25 μg/L in women and 10–20 μg/L in men. PRL secretion is pulsatile, with the highest secretory peaks occurring during rapid eye movement sleep. Peak serum PRL levels (up to 30 μg/L) occur between 4:00 and 6:00 A.M. The circulating half-life of PRL is about 50 min.
PRL is unique among the pituitary hormones in that the predominant central control mechanism is inhibitory, reflecting dopamine-mediated suppression of PRL release. This regulatory pathway accounts for the spontaneous PRL hypersecretion that occurs with pituitary stalk section, often a consequence of compressive mass lesions at the skull base. Pituitary dopamine type 2 (D2) receptors mediate inhibition of PRL synthesis and secretion. Targeted disruption (gene knockout) of the murine D2 receptor in mice results in hyperprolactinemia and lactotrope proliferation. As discussed below, dopamine agonists play a central role in the management of hyperprolactinemic disorders.
Thyrotropin-releasing hormone (TRH) (pyro Glu-His-Pro-NH2) is a hypothalamic tripeptide that elicits PRL release within 15–30 min after intravenous injection. The physiologic relevance of TRH for PRL regulation is unclear, and it appears primarily to regulate TSH (Chap. 405). Vasoactive intestinal peptide (VIP) also induces PRL release, whereas glucocorticoids and thyroid hormone weakly suppress PRL secretion.
Serum PRL levels rise transiently after exercise, meals, sexual intercourse, minor surgical procedures, general anesthesia, chest wall injury, acute myocardial infarction, and other forms of acute stress. PRL levels increase markedly (about tenfold) during pregnancy and decline rapidly within 2 weeks of parturition. If breast-feeding is initiated, basal PRL levels remain elevated; suckling stimulates transient reflex increases in PRL levels that last for about 30–45 min. Breast suckling activates neural afferent pathways in the hypothalamus that induce PRL release. With time, suckling-induced responses diminish and interfeeding PRL levels return to normal.
Action The PRL receptor is a member of the type I cytokine receptor family that also includes GH and interleukin (IL) 6 receptors. Ligand binding induces receptor dimerization and intracellular signaling by Janus kinase (JAK), which stimulates translocation of the signal transduction and activators of transcription (STAT) family to activate target genes. In the breast, the lobuloalveolar epithelium proliferates in response to PRL, placental lactogens, estrogen, progesterone, and local paracrine growth factors, including insulin-like growth factor I (IGF-I).
PRL acts to induce and maintain lactation, decrease reproductive function, and suppress sexual drive. These functions are geared toward ensuring that maternal lactation is sustained and not interrupted by pregnancy. PRL inhibits reproductive function by suppressing hypothalamic gonadotropin-releasing hormone (GnRH) and pituitary gonadotropin secretion and by impairing gonadal steroidogenesis in both women and men. In the ovary, PRL blocks folliculogenesis and inhibits granulosa cell aromatase activity, leading to hypoestrogenism and anovulation. PRL also has a luteolytic effect, generating a shortened, or inadequate, luteal phase of the menstrual cycle. In men, attenuated LH secretion leads to low testosterone levels and decreased spermatogenesis. These hormonal changes decrease libido and reduce fertility in patients with hyperprolactinemia.
GROWTH HORMONE
Synthesis GH is the most abundant anterior pituitary hormone, and GH-secreting somatotrope cells constitute up to 50% of the total anterior pituitary cell population. Mammosomatotrope cells, which coexpress PRL with GH, can be identified by using double immunostaining techniques. Somatotrope development and GH transcription are determined by expression of the cell-specific Pit-1 nuclear transcription factor. Five distinct genes encode GH and related proteins. The pituitary GH gene (hGH-N) produces two alternatively spliced products that give rise to 22-kDa GH (191 amino acids) and a less abundant 20-kDa GH molecule with similar biologic activity. Placental syncytiotrophoblast cells express a GH variant (hGH-V) gene; the related hormone human chorionic somatotropin (HCS) is expressed by distinct members of the gene cluster.
Secretion GH secretion is controlled by complex hypothalamic and peripheral factors. GH-releasing hormone (GHRH) is a 44-amino-acid hypothalamic peptide that stimulates GH synthesis and release. Ghrelin, an octanoylated gastric-derived peptide, and synthetic agonists of the GHS-R induce GHRH and also directly stimulate GH release. Somatostatin (somatotropin-release inhibiting factor [SRIF]) is synthesized in the medial preoptic area of the hypothalamus and inhibits GH secretion. GHRH is secreted in discrete spikes that elicit GH pulses, whereas SRIF sets basal GH secretory tone. SRIF also is expressed in many extrahypothalamic tissues, including the central nervous system (CNS), gastrointestinal tract, and pancreas, where it also acts to inhibit islet hormone secretion. IGF-I, the peripheral target hormone for GH, feeds back to inhibit GH; estrogen induces GH, whereas chronic glucocorticoid excess suppresses GH release.
Surface receptors on the somatotrope regulate GH synthesis and secretion. The GHRH receptor is a G protein–coupled receptor (GPCR) that signals through the intracellular cyclic AMP pathway to stimulate somatotrope cell proliferation as well as GH production. Inactivating mutations of the GHRH receptor cause profound dwarfism. A distinct surface receptor for ghrelin, the gastric-derived GH secretagogue, is expressed in both the hypothalamus and pituitary. Somatostatin binds to five distinct receptor subtypes (SSTR1 to SSTR5); SSTR2 and SSTR5 subtypes preferentially suppress GH (and TSH) secretion.
GH secretion is pulsatile, with highest peak levels occurring at night, generally correlating with sleep onset. GH secretory rates decline markedly with age so that hormone levels in middle age are about 15% of pubertal levels. These changes are paralleled by an age-related decline in lean muscle mass. GH secretion is also reduced in obese individuals, although IGF-I levels may not be suppressed, suggesting a change in the setpoint for feedback control. Elevated GH levels occur within an hour of deep sleep onset as well as after exercise, physical stress, and trauma and during sepsis. Integrated 24-h GH secretion is higher in women and is also enhanced by estrogen replacement likely reflective of increased peripheral GH-resistance. Using standard assays, random GH measurements are undetectable in ~50% of daytime samples obtained from healthy subjects and are also undetectable in most obese and elderly subjects. Thus, single random GH measurements do not distinguish patients with adult GH deficiency from normal persons.
GH secretion is profoundly influenced by nutritional factors. Using newer ultrasensitive GH assays with a sensitivity of 0.002 μg/L, a glucose load suppresses GH to <0.7 μg/L in women and to <0.07 μg/L in men. Increased GH pulse frequency and peak amplitudes occur with chronic malnutrition or prolonged fasting. GH is stimulated by intravenous L-arginine, dopamine, and apomorphine (a dopamine receptor agonist), as well as by α-adrenergic pathways. β-Adrenergic blockade induces basal GH and enhances GHRH- and insulin-evoked GH release.
Action The pattern of GH secretion may affect tissue responses. The higher GH pulsatility observed in men compared with the relatively continuous basal GH secretion in women may be an important biologic determinant of linear growth patterns and liver enzyme induction.
The 70-kDa peripheral GH receptor protein has structural homology with the cytokine/hematopoietic superfamily. A fragment of the receptor extracellular domain generates a soluble GH binding protein (GHBP) that interacts with GH in the circulation. The liver and cartilage contain the greatest number of GH receptors. GH binding to preformed receptor dimers is followed by internal rotation and subsequent signaling through the JAK/STAT pathway. Activated STAT proteins translocate to the nucleus, where they modulate expression of GH-regulated target genes. GH analogues that bind to the receptor but are incapable of mediating receptor signaling are potent antagonists of GH action. A GH receptor antagonist (pegvisomant) is approved for treatment of acromegaly.
GH induces protein synthesis and nitrogen retention and impairs glucose tolerance by antagonizing insulin action. GH also stimulates lipolysis, leading to increased circulating fatty acid levels, reduced omental fat mass, and enhanced lean body mass. GH promotes sodium, potassium, and water retention and elevates serum levels of inorganic phosphate. Linear bone growth occurs as a result of complex hormonal and growth factor actions, including those of IGF-I. GH stimulates epiphyseal prechondrocyte differentiation. These precursor cells produce IGF-I locally, and their proliferation is also responsive to the growth factor.
Insulin-Like Growth Factors Although GH exerts direct effects in target tissues, many of its physiologic effects are mediated indirectly through IGF-I, a potent growth and differentiation factor. The liver is the major source of circulating IGF-I. In peripheral tissues, IGF-I also exerts local paracrine actions that appear to be both dependent on and independent of GH. Thus, GH administration induces circulating IGF-I as well as stimulating local IGF-I production in multiple tissues.
Both IGF-I and IGF-II are bound to high-affinity circulating IGF-binding proteins (IGFBPs) that regulate IGF bioactivity. Levels of IGFBP3 are GH-dependent, and it serves as the major carrier protein for circulating IGF-I. GH deficiency and malnutrition usually are associated with low IGFBP3 levels. IGFBP1 and IGFBP2 regulate local tissue IGF action but do not bind appreciable amounts of circulating IGF-I.
Serum IGF-I concentrations are profoundly affected by physiologic factors. Levels increase during puberty, peak at 16 years, and subsequently decline by >80% during the aging process. IGF-I concentrations are higher in women than in men. Because GH is the major determinant of hepatic IGF-I synthesis, abnormalities of GH synthesis or action (e.g., pituitary failure, GHRH receptor defect, GH receptor defect or pharmacologic GH receptor blockade) reduce IGF-I levels. Hypocaloric states are associated with GH resistance; IGF-I levels are therefore low with cachexia, malnutrition, and sepsis. In acromegaly, IGF-I levels are invariably high and reflect a log-linear relationship with circulating GH concentrations.
IGF-I PHYSIOLOGY Injected IGF-I (100 μg/kg) induces hypoglycemia, and lower doses improve insulin sensitivity in patients with severe insulin resistance and diabetes. In cachectic subjects, IGF-I infusion (12 μg/kg per hour) enhances nitrogen retention and lowers cholesterol levels. Longer-term subcutaneous IGF-I injections enhance protein synthesis and are anabolic. Although bone formation markers are induced, bone turnover also may be stimulated by IGF-I. IGF-I has only been approved for use in patients with GH-resistance syndromes.
IGF-I side effects are dose-dependent, and overdose may result in hypoglycemia, hypotension, fluid retention, temporomandibular jaw pain, and increased intracranial pressure, all of which are reversible. Avascular femoral head necrosis has been reported. Chronic excess IGF-I administration presumably would result in features of acromegaly.
ADRENOCORTICOTROPIC HORMONE
(See also Chap. 406)
Synthesis ACTH-secreting corticotrope cells constitute about 20% of the pituitary cell population. ACTH (39 amino acids) is derived from the POMC precursor protein (266 amino acids) that also generates several other peptides, including β-lipotropin, β-endorphin, met-enkephalin, α-melanocyte-stimulating hormone (α-MSH), and corticotropin-like intermediate lobe protein (CLIP). The POMC gene is potently suppressed by glucocorticoids and induced by corticotropin-releasing hormone (CRH), arginine vasopressin (AVP), and proinflammatory cytokines, including IL-6, as well as leukemia inhibitory factor.
CRH, a 41-amino-acid hypothalamic peptide synthesized in the paraventricular nucleus as well as in higher brain centers, is the predominant stimulator of ACTH synthesis and release. The CRH receptor is a GPCR that is expressed on the corticotrope and signals to induce POMC transcription.
Secretion ACTH secretion is pulsatile and exhibits a characteristic circadian rhythm, peaking at about 6 A.M. and reaching a nadir about midnight. Adrenal glucocorticoid secretion, which is driven by ACTH, follows a parallel diurnal pattern. ACTH circadian rhythmicity is determined by variations in secretory pulse amplitude rather than changes in pulse frequency. Superimposed on this endogenous rhythm, ACTH levels are increased by physical and psychological stress, exercise, acute illness, and insulin-induced hypoglycemia.
Glucocorticoid-mediated negative regulation of the hypothalamic-pituitary-adrenal (HPA) axis occurs as a consequence of both hypothalamic CRH suppression and direct attenuation of pituitary POMC gene expression and ACTH release. In contrast, loss of cortisol feedback inhibition, as occurs in primary adrenal failure, results in extremely high ACTH levels.
Acute inflammatory or septic insults activate the HPA axis through the integrated actions of proinflammatory cytokines, bacterial toxins, and neural signals. The overlapping cascade of ACTH-inducing cytokines (tumor necrosis factor [TNF]; IL-1, -2, and -6; and leukemia inhibitory factor) activates hypothalamic CRH and AVP secretion, pituitary POMC gene expression, and local pituitary paracrine cytokine networks. The resulting cortisol elevation restrains the inflammatory response and enables host protection. Concomitantly, cytokine-mediated central glucocorticoid receptor resistance impairs glucocorticoid suppression of the HPA. Thus, the neuroendocrine stress response reflects the net result of highly integrated hypothalamic, intrapituitary, and peripheral hormone and cytokine signals acting to regulate cortisol secretion.
Action The major function of the HPA axis is to maintain metabolic homeostasis and mediate the neuroendocrine stress response. ACTH induces adrenocortical steroidogenesis by sustaining adrenal cell proliferation and function. The receptor for ACTH, designated melanocortin-2 receptor, is a GPCR that induces steroidogenesis by stimulating a cascade of steroidogenic enzymes (Chap. 406).
GONADOTROPINS: FSH AND LH
Synthesis and Secretion Gonadotrope cells constitute about 10% of anterior pituitary cells and produce two gonadotropin hormones—LH and FSH. Like TSH and hCG, LH and FSH are glycoprotein hormones that comprise α and β subunits. The α subunit is common to these glycoprotein hormones; specificity of hormone function is conferred by the β subunits, which are expressed by separate genes.
Gonadotropin synthesis and release are dynamically regulated. This is particularly true in women, in whom rapidly fluctuating gonadal steroid levels vary throughout the menstrual cycle. Hypothalamic GnRH, a 10-amino-acid peptide, regulates the synthesis and secretion of both LH and FSH. Brain kisspeptin, a product of the KISSI gene regulates hypothalamic GnRH release. GnRH is secreted in discrete pulses every 60–120 min, and the pulses in turn elicit LH and FSH pulses (Fig. 401e-3). The pulsatile mode of GnRH input is essential to its action; pulses prime gonadotrope responsiveness, whereas continuous GnRH exposure induces desensitization. Based on this phenomenon, long-acting GnRH agonists are used to suppress gonadotropin levels in children with precocious puberty and in men with prostate cancer (Chap. 115) and are used in some ovulation-induction protocols to reduce levels of endogenous gonadotropins (Chap. 412). Estrogens act at both the hypothalamus and the pituitary to modulate gonadotropin secretion. Chronic estrogen exposure is inhibitory, whereas rising estrogen levels, as occur during the preovulatory surge, exert positive feedback to increase gonadotropin pulse frequency and amplitude. Progesterone slows GnRH pulse frequency but enhances gonadotropin responses to GnRH. Testosterone feedback in men also occurs at the hypothalamic and pituitary levels and is mediated in part by its conversion to estrogens.
Although GnRH is the main regulator of LH and FSH secretion, FSH synthesis is also under separate control by the gonadal peptides inhibin and activin, which are members of the transforming growth factor β (TGF-β) family. Inhibin selectively suppresses FSH, whereas activin stimulates FSH synthesis (Chap. 412).
Action The gonadotropin hormones interact with their respective GPCRs expressed in the ovary and testis, evoking germ cell development and maturation and steroid hormone biosynthesis. In women, FSH regulates ovarian follicle development and stimulates ovarian estrogen production. LH mediates ovulation and maintenance of the corpus luteum. In men, LH induces Leydig cell testosterone synthesis and secretion, and FSH stimulates seminiferous tubule development and regulates spermatogenesis.
THYROID-STIMULATING HORMONE
Synthesis and Secretion TSH-secreting thyrotrope cells constitute 5% of the anterior pituitary cell population. TSH shares a common α subunit with LH and FSH but contains a specific TSH β subunit. TRH is a hypothalamic tripeptide (pyroglutamyl histidylprolinamide) that acts through a pituitary GPCR to stimulate TSH synthesis and secretion; it also stimulates the lactotrope cell to secrete PRL. TSH secretion is stimulated by TRH, whereas thyroid hormones, dopamine, somatostatin, and glucocorticoids suppress TSH by overriding TRH induction.
Thyrotrope cell proliferation and TSH secretion are both induced when negative feedback inhibition by thyroid hormones is removed. Thus, thyroid damage (including surgical thyroidectomy), radiation-induced hypothyroidism, chronic thyroiditis, and prolonged goitrogen exposure are associated with increased TSH levels. Long-standing untreated hypothyroidism can lead to elevated TSH levels as well as thyrotrope hyperplasia and pituitary enlargement, which may be evident on magnetic resonance imaging.
Action TSH is secreted in pulses, although the excursions are modest in comparison to other pituitary hormones because of the low amplitude of the pulses and the relatively long half-life of TSH. Consequently, single determinations of TSH suffice to precisely assess its circulating levels. TSH binds to a GPCR on thyroid follicular cells to stimulate thyroid hormone synthesis and release (Chap. 405).
402 | Hypopituitarism |
Inadequate production of anterior pituitary hormones leads to features of hypopituitarism. Impaired production of one or more of the anterior pituitary trophic hormones can result from inherited disorders; more commonly, adult hypopituitarism is acquired and reflects the compressive mass effects of tumors or the consequences of local pituitary or hypothalamic traumatic, inflammatory, or vascular damage. These processes also may impair synthesis or secretion of hypothalamic hormones, with resultant pituitary failure (Table 402-1).
ETIOLOGY OF HYPOPITUITARISMa |



