Hypertension
KEY CONCEPTS
![]() The risk of cardiovascular (CV) morbidity and mortality is directly correlated with blood pressure (BP).
The risk of cardiovascular (CV) morbidity and mortality is directly correlated with blood pressure (BP).
![]() Evidence from clinical trials have shown that antihypertensive drug therapy substantially reduces the risks of CV events and death in patients with high BP.
Evidence from clinical trials have shown that antihypertensive drug therapy substantially reduces the risks of CV events and death in patients with high BP.
![]() Essential hypertension is usually an asymptomatic disease. A diagnosis cannot be made based on one elevated BP measurement. An elevated value from the average of two or more measurements, present during two or more clinical encounters, is needed to diagnose hypertension.
Essential hypertension is usually an asymptomatic disease. A diagnosis cannot be made based on one elevated BP measurement. An elevated value from the average of two or more measurements, present during two or more clinical encounters, is needed to diagnose hypertension.
![]() The overall goal of treating hypertension is to reduce hypertension-associated morbidity and mortality from CV events. These are considered hypertension-associated complications. The selection of specific drug therapy should be based on evidence that demonstrates CV risk reduction.
The overall goal of treating hypertension is to reduce hypertension-associated morbidity and mortality from CV events. These are considered hypertension-associated complications. The selection of specific drug therapy should be based on evidence that demonstrates CV risk reduction.
![]() A goal BP of <140/90 mm Hg is appropriate for general prevention of CV events and CV risk reduction in most patients. For some patients (e.g., diabetes and/or significant chronic kidney disease) lower goal BP values may be appropriate on a patient-specific basis.
A goal BP of <140/90 mm Hg is appropriate for general prevention of CV events and CV risk reduction in most patients. For some patients (e.g., diabetes and/or significant chronic kidney disease) lower goal BP values may be appropriate on a patient-specific basis.
![]() Magnitude of BP elevation should be used to guide determination of the number of agents to start when implementing drug therapy. Most patients with stage 1 hypertension should be started on one drug, with the option of starting two for some patients. However, most patients presenting with stage 2 hypertension should be started on two drugs.
Magnitude of BP elevation should be used to guide determination of the number of agents to start when implementing drug therapy. Most patients with stage 1 hypertension should be started on one drug, with the option of starting two for some patients. However, most patients presenting with stage 2 hypertension should be started on two drugs.
![]() Lifestyle modifications should be prescribed in all patients, especially those with prehypertension and hypertension. However, they should never be used as a replacement for antihypertensive drug therapy for patients with hypertension, especially in those with additional CV risk factors.
Lifestyle modifications should be prescribed in all patients, especially those with prehypertension and hypertension. However, they should never be used as a replacement for antihypertensive drug therapy for patients with hypertension, especially in those with additional CV risk factors.
![]() Angiotensin-converting enzyme (ACE) inhibitors, angiotensin II receptor blockers (ARBs), calcium channel blockers (CCBs), and thiazide diuretics are all first-line agents for most patients with hypertension for general prevention of CV events and CV risk reduction. These first-line options are for patients with hypertension who do not have any compelling indications for a specific antihypertensive drug class.
Angiotensin-converting enzyme (ACE) inhibitors, angiotensin II receptor blockers (ARBs), calcium channel blockers (CCBs), and thiazide diuretics are all first-line agents for most patients with hypertension for general prevention of CV events and CV risk reduction. These first-line options are for patients with hypertension who do not have any compelling indications for a specific antihypertensive drug class.
![]() For general prevention of CV events and CV risk reduction in most patients with hypertension, β-blockers do not reduce CV events to the extent that has been proven with thiazide-type diuretics, ACE inhibitors, ARBs, CCBs, or thiazide diuretics.
For general prevention of CV events and CV risk reduction in most patients with hypertension, β-blockers do not reduce CV events to the extent that has been proven with thiazide-type diuretics, ACE inhibitors, ARBs, CCBs, or thiazide diuretics.
![]() Compelling indications are comorbid conditions where specific antihypertensive drug classes have been shown in clinical trials to provide unique long-term benefits (reducing the risk of CV events).
Compelling indications are comorbid conditions where specific antihypertensive drug classes have been shown in clinical trials to provide unique long-term benefits (reducing the risk of CV events).
![]() Patients with diabetes are at high risk for CV events. All patients with diabetes and hypertension should ideally be managed with either an ACE inhibitor or an ARB. These are typically in combination with one or more other antihypertensive agents because multiple agents frequently are needed to control BP.
Patients with diabetes are at high risk for CV events. All patients with diabetes and hypertension should ideally be managed with either an ACE inhibitor or an ARB. These are typically in combination with one or more other antihypertensive agents because multiple agents frequently are needed to control BP.
![]() Older patients are often at risk for orthostatic hypotension when antihypertensive drug therapy is started. Although overall antihypertensive drug therapy should be the same as in younger patients, low initial doses should be used and dosage titrations should be gradual to minimize risk of orthostatic hypotension.
Older patients are often at risk for orthostatic hypotension when antihypertensive drug therapy is started. Although overall antihypertensive drug therapy should be the same as in younger patients, low initial doses should be used and dosage titrations should be gradual to minimize risk of orthostatic hypotension.
![]() Alternative antihypertensive agents have not been proven to reduce the risk of CV events to the same extent compared with first-line antihypertensive agents. They should be used primarily in combination with first-line agents to provide additional BP lowering.
Alternative antihypertensive agents have not been proven to reduce the risk of CV events to the same extent compared with first-line antihypertensive agents. They should be used primarily in combination with first-line agents to provide additional BP lowering.
![]() Initial therapy with the combination of two antihypertensive agents should be used in most patients presenting with stage 2 hypertension. This is also an option for patients presenting with stage 1 hypertension. Most patients require combination therapy to achieve goal BP.
Initial therapy with the combination of two antihypertensive agents should be used in most patients presenting with stage 2 hypertension. This is also an option for patients presenting with stage 1 hypertension. Most patients require combination therapy to achieve goal BP.
![]() Patients have resistant hypertension when they fail to attain goal BP values while adherent to a regimen that includes at least three agents at maximum dose, one of which includes a diuretic, or when four or more agents are needed to treat hypertension.
Patients have resistant hypertension when they fail to attain goal BP values while adherent to a regimen that includes at least three agents at maximum dose, one of which includes a diuretic, or when four or more agents are needed to treat hypertension.
![]() Hypertensive urgency is ideally managed by adjusting maintenance therapy, adding a new antihypertensive, and/or increasing the dose of a present medication. This provides a gradual reduction in BP, which is a safer treatment approach than rapid reductions in BP.
Hypertensive urgency is ideally managed by adjusting maintenance therapy, adding a new antihypertensive, and/or increasing the dose of a present medication. This provides a gradual reduction in BP, which is a safer treatment approach than rapid reductions in BP.
Hypertension is a common disease that is simply defined as persistently elevated arterial blood pressure (BP). Although elevated BP was perceived to be “essential” for adequate perfusion of vital organs during the early and middle 1900s, it is now identified as one of the most significant risk factors for cardiovascular (CV) disease. Increasing awareness and diagnosis of hypertension, and improving control of BP with appropriate treatment are considered critical public health initiatives to reduce CV morbidity and mortality.
The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC7) is the most prominent evidence-based clinical guideline in the United States for the management of hypertension.1 The Eighth Report of the Joint National Committee was under development for several years. In 2013, the sponsoring organization (National Heart Lung and Blood Institute [NHLBI]) decided that the work of the JNC8 groups will be published as evidentiary reviews, not a guideline. However, the NHLBI will subsequently collaborate with another organization to prepare and issue an updated hypertension clinical practice guideline sometime after 2013. Once available, this will likely be viewed as the preeminent hypertension clinical guideline in the United States. The 2007 American Heart Association (AHA) Scientific Statement on the treatment of hypertension provides additional insight regarding pharmacotherapy for hypertension.2 This chapter reviews relevant components of both these consensus documents and additional evidence from clinical trials, with a focus on the pharmacotherapy of hypertension.
The National Health and Nutrition Examination Survey and the National Center for Health Statistics regularly assess hypertension in the United States.3 Data from 2007 to 2010 indicate that 77.9 million Americans aged 20 years and above have hypertension. Predictions indicate that by 2030 the prevalence of hypertension will increase by 7.2%. Approximately half of all patients with hypertension are at their goal BP value. While this control rate is substantially higher than in the past, there remain many opportunities for clinicians to improve the care of patients with hypertension.
EPIDEMIOLOGY
Approximately 1 in 3 adult Americans (77.9 million people) have elevated BP, defined as ≥140/90 mm Hg.3 The overall incidence is similar between men and women, but varies depending on age. The percentage of men with high BP is higher than that of women before the age of 55 and is similar to that of women between the ages 55 and 64. However, after the age of 64, a much higher percentage of women have high BP than men.3 Prevalence rates are highest in non-Hispanic blacks (47% in women, 43% in men), followed by non-Hispanic whites (31% in women, 33% in men), and Mexican Americans (29% in women, 30% in men).3 With regard to BP control, 54.9% of non-Hispanic whites have controlled hypertension in contrast to 47.6% of non-Hispanic blacks and 39.3% in Mexican Americans.3
BP values increase with age, and hypertension (persistently elevated BP values) is very common in the elderly. The lifetime risk of developing hypertension among those 55 years of age and older who are normotensive is 90%.1 Most patients have prehypertension before they are diagnosed with hypertension, with most diagnoses occurring between the third and fifth decades of life.
ETIOLOGY
In most patients, hypertension results from unknown pathophysiologic etiology (essential or primary hypertension). This form of hypertension cannot be cured, but it can be controlled. A small percentage of patients have a specific cause of their hypertension (secondary hypertension). There are many potential secondary causes that either are concurrent medical conditions or are endogenously induced. If the cause can be identified, hypertension in these patients can be mitigated or potentially be cured.
Essential Hypertension
Over 90% of individuals with high BP have essential hypertension.1 Numerous mechanisms have been identified that may contribute to the pathogenesis of this form of hypertension, so identifying the exact underlying abnormality is not possible. Genetic factors may play an important role in the development of essential hypertension. There are monogenic and polygenic forms of BP dysregulation that may be responsible for essential hypertension.4 Many of these genetic traits feature genes that affect sodium balance, but genetic mutations altering urinary kallikrein excretion, nitric oxide release, and excretion of aldosterone, other adrenal steroids, and angiotensinogen are also documented.4 In the future, genetic testing for these traits could lead to alternative approaches to preventing or treating hypertension; however, this is not currently recommended.
Secondary Hypertension
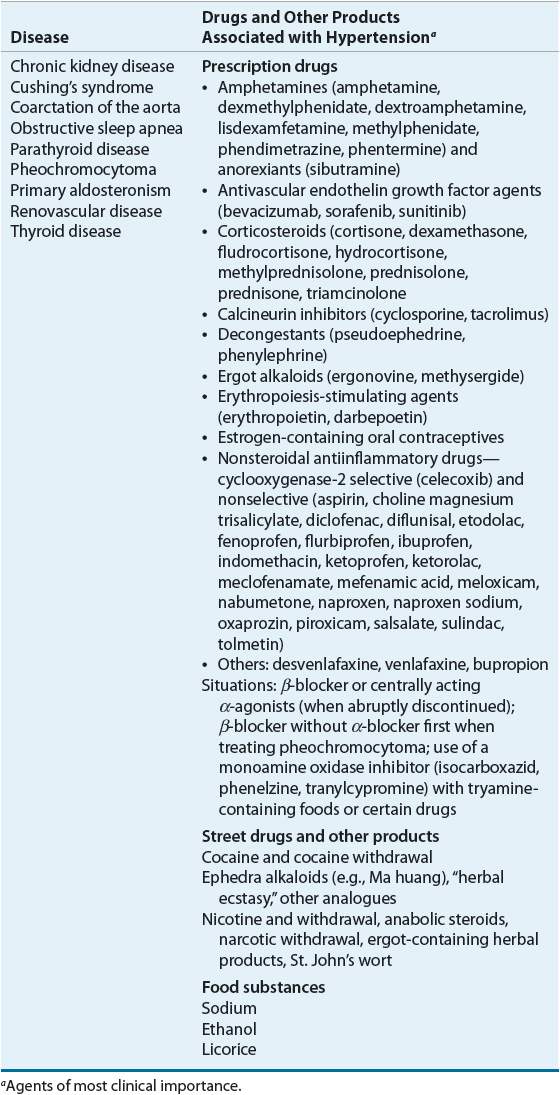
Fewer than 10% of patients have secondary hypertension where either a comorbid disease or a drug (or other product) is responsible for elevating BP (see Table 3-1).1,5 In most of these cases, renal dysfunction resulting from severe chronic kidney disease (CKD) or renovascular disease is the most common secondary cause. Certain drugs (or other products), either directly or indirectly, can cause hypertension or exacerbate hypertension by increasing BP. The most common agents are listed in Table 3-1. When a secondary cause is identified, removing the offending agent (when feasible) or treating/correcting the underlying comorbid condition should be the first step in management.
TABLE 3-1 Secondary Causes of Hypertension
PATHOPHYSIOLOGY
Multiple factors that control BP are potential contributing components in the development of essential hypertension.4,6 These include malfunctions in either humoral (i.e., the renin–angiotensin–aldosterone system [RAAS]) or vasodepressor mechanisms, abnormal neuronal mechanisms, defects in peripheral autoregulation, and disturbances in sodium, calcium, and natriuretic hormone. Many of these factors are cumulatively affected by the multifaceted RAAS, which ultimately regulates arterial BP. It is probable that no one factor is solely responsible for essential hypertension.
Arterial BP
Arterial BP is the pressure in the arterial wall measured in millimeters of mercury (mm Hg). The two typical arterial BP values are systolic BP (SBP) and diastolic BP (DBP). SBP represents the peak value, which is achieved during cardiac contraction. DBP is achieved after contraction when the cardiac chambers are filling, and represents the nadir value. The difference between SBP and DBP is called the pulse pressure and is a measure of arterial wall tension. Mean arterial pressure (MAP) is the average pressure throughout the cardiac cycle of contraction. It is sometimes used clinically to represent overall arterial BP, especially in hypertensive emergency. During a cardiac cycle, two thirds of the time is spent in diastole and one third in systole. Therefore, the MAP is calculated by using the following equation:
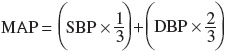
Arterial BP is hemodynamically generated by the interplay between blood flow and the resistance to blood flow. It is mathematically defined as the product of cardiac output (CO) and total peripheral resistance (TPR) according to the following equation:
![]()
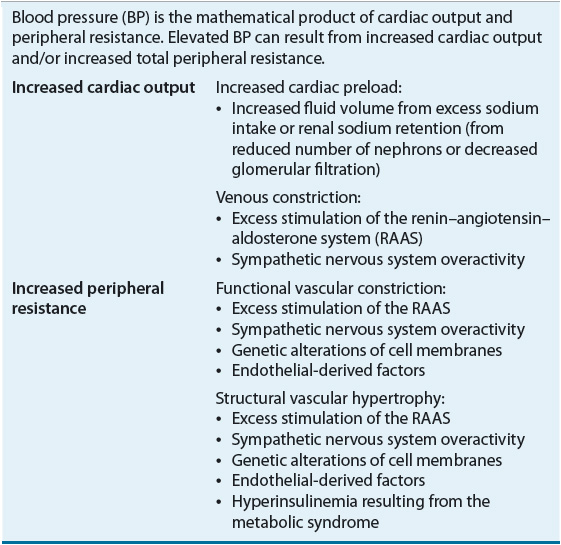
CO is the major determinant of SBP, whereas TPR largely determines DBP. In turn, CO is a function of stroke volume, heart rate, and venous capacitance. Table 3-2 lists physiologic causes of increased CO and TPR and correlates them to potential mechanisms of pathogenesis.
TABLE 3-2 Potential Mechanisms of Pathogenesis
Under normal physiologic conditions, arterial BP fluctuates throughout the day following a circadian rhythm. BP decreases to its lowest daily values during sleep followed by a sharp rise starting a few hours prior to awakening with the highest values occurring midmorning. BP is also increased acutely during physical activity or emotional stress.
Classification
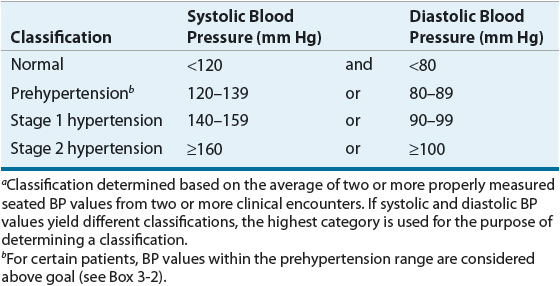
The classification of BP in adults (age 18 years and older) is based on the average of two or more properly measured BP values from two or more clinical encounters (Table 3-3).1 It includes four categories: normal, prehypertension, stage 1 hypertension, and stage 2 hypertension. Prehypertension is not considered a disease category but identifies patients whose BP is likely to increase into the classification of hypertension in the future.
TABLE 3-3 Classification of Blood Pressure in Adults (Age ≥18 Years)a
Hypertensive crises are clinical situations where BP values are very elevated, typically >180/120 mm Hg.6 They are categorized as either hypertensive emergency or hypertensive urgency. Hypertensive emergencies are extreme elevations in BP that are accompanied by acute or progressing target-organ damage. Hypertensive urgencies are high elevations in BP without acute or progressing target-organ injury.
Cardiovascular Risk and Blood Pressure
![]() Epidemiologic data demonstrate a strong correlation between BP and CV morbidity and mortality.7 Risk of stroke, myocardial infarction (MI), angina, heart failure, kidney failure, or early death from a CV cause is directly correlated with BP. Starting at a BP of 115/75 mm Hg, risk of CV disease doubles with every 20/10 mm Hg increase.1 Even patients with prehypertension have an increased risk of CV disease.
Epidemiologic data demonstrate a strong correlation between BP and CV morbidity and mortality.7 Risk of stroke, myocardial infarction (MI), angina, heart failure, kidney failure, or early death from a CV cause is directly correlated with BP. Starting at a BP of 115/75 mm Hg, risk of CV disease doubles with every 20/10 mm Hg increase.1 Even patients with prehypertension have an increased risk of CV disease.
![]() Treating patients with hypertension with antihypertensive drug therapy provides significant clinical benefits. Evidence from large-scale placebo-controlled clinical trials have shown that the increased risks of CV events and death associated with elevated BP are reduced substantially by antihypertensive therapy.8–11 This is discussed in Treatment section of this chapter.
Treating patients with hypertension with antihypertensive drug therapy provides significant clinical benefits. Evidence from large-scale placebo-controlled clinical trials have shown that the increased risks of CV events and death associated with elevated BP are reduced substantially by antihypertensive therapy.8–11 This is discussed in Treatment section of this chapter.
SBP is a stronger predictor of CV disease than DBP in adults aged 50 years and older; it is the most important clinical BP parameter for most patients.1 Patients are considered to have isolated systolic hypertension when their SBP values are elevated (i.e., ≥140 mm Hg) and DBP values are not (i.e., <90 mm Hg, but commonly <80 mm Hg). Isolated systolic hypertension is believed to result from pathophysiologic changes in the arterial vasculature consistent with aging. These changes decrease the compliance of the arterial wall and portend an increased risk of CV morbidity and mortality. The elevated pulse pressure (SBP minus DBP) is believed to reflect extent of atherosclerotic disease in the elderly and is a measure of increased arterial stiffness. Higher pulse pressure values seen in those with isolated systolic hypertension are directly correlated with risk of CV mortality.
Humoral Mechanisms
Several humoral abnormalities involving the RAAS, natriuretic hormone, and hyperinsulinemia may be involved in the development of essential hypertension.
The Renin–Angiotensin–Aldosterone System
The RAAS is a complex endogenous system involved with most regulatory components of arterial BP. Activation and regulation is primarily governed by the kidney (see Fig. 3-1). The RAAS regulates sodium, potassium, and blood volume. Therefore, this system significantly influences vascular tone and sympathetic nervous system activity, and is the most influential contributor to the homeostatic regulation of BP.
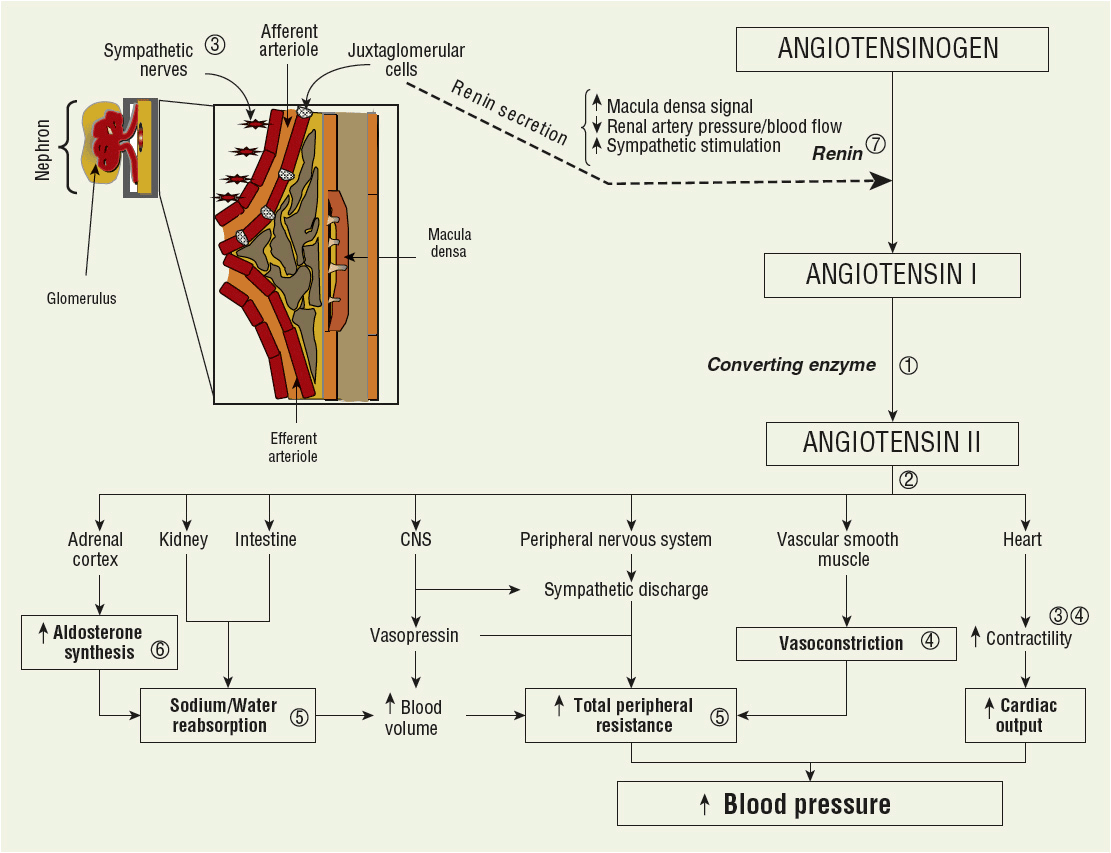
FIGURE 3-1 Diagram representing the renin–angiotensin–aldosterone system. The interrelationship between the kidney, angiotensin II, and regulation of blood pressure is depicted. Renin secretion from the juxtaglomerular cells in the afferent arterioles is regulated by three major factors to trigger conversion of angiotensinogen to angiotensin 1. The primary sites of action for major antihypertensive agents are included: ![]() ACE inhibitors;
ACE inhibitors; ![]() angiotensin II receptor blockers;
angiotensin II receptor blockers; ![]() β-blockers;
β-blockers; ![]() calcium channel blockers;
calcium channel blockers; ![]() diuretics;
diuretics; ![]() aldosterone antagonists;
aldosterone antagonists; ![]() direct renin inhibitor.
direct renin inhibitor.
Renin is an enzyme that is stored in the juxtaglomerular cells, which are located in the afferent arterioles of the kidney. The release of renin is modulated by several factors: intrarenal factors (e.g., renal perfusion pressure, catecholamines, angiotensin II) and extrarenal factors (e.g., sodium, chloride, and potassium).
Juxtaglomerular cells function as a baroreceptor-sensing device. Decreased renal artery pressure and kidney blood flow is sensed by these cells and stimulates secretion of renin. The juxtaglomerular apparatus also includes a group of specialized distal tubule cells referred to collectively as the macula densa. A decrease in sodium and chloride delivered to the distal tubule stimulates renin release. Catecholamines increase renin release probably by directly stimulating sympathetic nerves on the afferent arterioles that in turn activate the juxtaglomerular cells.
Renin catalyzes the conversion of angiotensinogen to angiotensin I in the blood. Angiotensin I is then converted to angiotensin II by angiotensin-converting enzyme (ACE). After binding to specific receptors (classified as either angiotensin II type 1 [AT1] or angiotensin II type 2 [AT2] subtypes), angiotensin II exerts biologic effects in several tissues. The AT1 receptor is located in brain, kidney, myocardium, peripheral vasculature, and the adrenal glands. These receptors mediate most responses that are critical to CV and kidney function. The AT2 receptor is located in adrenal medullary tissue, uterus, and brain. Stimulation of the AT2 receptor does not influence BP regulation.
Circulating angiotensin II can elevate BP through pressor and volume effects. Pressor effects include direct vasoconstriction, stimulation of catecholamine release from the adrenal medulla, and centrally mediated increases in sympathetic nervous system activity. Angiotensin II also stimulates aldosterone synthesis from the adrenal cortex. This leads to sodium and water reabsorption that increases plasma volume, TPR, and ultimately BP. Aldosterone also has a deleterious role in the pathophysiology of other CV diseases (e.g., heart failure, MI, kidney disease) by promoting tissue remodeling leading to myocardial fibrosis and vascular dysfunction. Clearly, any disturbance in the body that leads to activation of the RAAS could explain chronic hypertension.
The heart and brain contain a local RAAS. In the heart, angiotensin II is also generated by angiotensin I convertase (human chymase). This enzyme is not blocked by ACE inhibition. Activation of the myocardial RAAS increases cardiac contractility and stimulates cardiac hypertrophy. In the brain, angiotensin II modulates the production and release of hypothalamic and pituitary hormones, and enhances sympathetic outflow from the medulla oblongata.
Natriuretic Hormone
Natriuretic hormone inhibits sodium and potassium-ATPase and thus interferes with sodium transport across cell membranes. Inherited defects in the kidney’s ability to eliminate sodium can cause increased blood volume. A compensatory increase in the concentration of circulating natriuretic hormone theoretically could increase urinary excretion of sodium and water. However, this hormone might block the active transport of sodium out of arteriolar smooth muscle cells. The increased intracellular sodium concentration ultimately would increase vascular tone and BP.
Insulin Resistance and Hyperinsulinemia
The combination of multiple CV and metabolic abnormalities is referred to as the metabolic syndrome.12 Hypothetically, increased insulin concentrations may lead to hypertension because of increased renal sodium retention and enhanced sympathetic nervous system activity. Moreover, insulin has growth hormone–like actions that can induce hypertrophy of vascular smooth muscle cells. Insulin also may elevate BP by increasing intracellular calcium, which leads to increased vascular resistance. The exact mechanism by which insulin resistance and hyperinsulinemia occur in hypertension is unknown. However, this association is strong because many of the criteria used to define this population (i.e., elevated BP, abdominal obesity, high triglycerides, low high-density lipoprotein cholesterol, and elevated fasting glucose) are often present in patients with hypertension.12
Neuronal Regulation
Central and autonomic nervous systems are intricately involved in the regulation of arterial BP. Many receptors that either enhance or inhibit norepinephrine release are located on the presynaptic surface of sympathetic terminals. The α and β presynaptic receptors play a role in negative and positive feedback to the norepinephrine-containing vesicles. Stimulation of presynaptic α-receptors (α2) exerts a negative inhibition on norepinephrine release. Stimulation of presynaptic β-receptors facilitates norepinephrine release.
Sympathetic neuronal fibers located on the surface of effector cells innervate the α– and β-receptors. Stimulation of postsynaptic α-receptors (α1) on arterioles and venules results in vasoconstriction. There are two types of postsynaptic β-receptors, β1 and β2. Both are present in all tissues innervated by the sympathetic nervous system. However, in some tissues β1-receptors predominate (e.g., heart), and in other tissues β2-receptors predominate (e.g., bronchioles). Stimulation of β1-receptors in the heart results in an increase in heart rate (chronotropy) and force of contraction (ionotropy), whereas stimulation of β2-receptors in the arterioles and venules causes vasodilation.
The baroreceptor reflex system is the major negative feedback mechanism that controls sympathetic activity. Baroreceptors are nerve endings lying in the walls of large arteries, especially in the carotid arteries and aortic arch. Changes in arterial BP rapidly activate baroreceptors that then transmit impulses to the brain stem through the ninth cranial nerve and vagus nerve. In this reflex system, a decrease in arterial BP stimulates baroreceptors, causing reflex vasoconstriction and increased heart rate and force of cardiac contraction. These baroreceptor reflex mechanisms may be less responsive in the elderly and those with diabetes.
Stimulation of certain areas within the central nervous system (e.g., nucleus tractus solitarius, vagal nuclei, vasomotor center, area postrema) can either increase or decrease BP. For example, α2-adrenergic stimulation within the central nervous system decreases BP through an inhibitory effect on the vasomotor center. However, angiotensin II increases sympathetic outflow from the vasomotor center, which increases BP.
The purpose of these neuronal mechanisms is to regulate BP and maintain homeostasis. Pathologic disturbances in any of the four major components (autonomic nerve fibers, adrenergic receptors, baroreceptors, central nervous system) could chronically elevate BP. These systems are physiologically interrelated. A defect in one component may alter normal function in another. Therefore, cumulative abnormalities may explain the development of essential hypertension.
Peripheral Autoregulatory Components
Abnormalities in renal or tissue autoregulatory systems could cause hypertension. It is possible that a renal defect in sodium excretion may develop, which can then cause resetting of tissue autoregulatory processes resulting in a higher BP. The kidney usually maintains a normal BP through a volume–pressure adaptive mechanism. When BP drops, the kidneys respond by increasing retention of sodium and water, which leads to plasma volume expansion that increases BP. Conversely, when BP rises above normal, renal sodium and water excretion are increased to reduce plasma volume and CO.
Local autoregulatory processes maintain adequate tissue oxygenation. When tissue oxygen demand is normal to low, the local arteriolar bed remains relatively vasoconstricted. However, increase in metabolic demand triggers arteriolar vasodilation that lowers peripheral vascular resistance (PVR) and increases blood flow and oxygen delivery.
Intrinsic defects in renal adaptive mechanisms could lead to plasma volume expansion and increased blood flow to peripheral tissues, even when BP is normal. Local tissue autoregulatory processes that vasoconstrict would then be activated to offset the increased blood flow. This effect would result in increased PVR and, if sustained, would also result in thickening of the arteriolar walls. This pathophysiologic component is plausible because increased TPR is a common underlying finding in patients with essential hypertension.
Vascular Endothelial Mechanisms
Vascular endothelium and smooth muscle play important roles in regulating blood vessel tone and BP. These regulating functions are mediated by vasoactive substances that are synthesized by endothelial cells. It has been postulated that a deficiency in local synthesis of vasodilating substances (e.g., prostacyclin and bradykinin) or excess vasoconstricting substances (e.g., angiotensin II and endothelin I) contribute to essential hypertension, atherosclerosis, and other CV diseases.
Nitric oxide is produced in the endothelium, relaxes the vascular epithelium, and is a very potent vasodilator. The nitric oxide system is an important regulator of arterial BP. Patients with hypertension may have an intrinsic nitric oxide deficiency, resulting in inadequate vasodilation.
Electrolytes
Epidemiologic and clinical data have associated excess sodium intake with hypertension. Population-based studies indicate that high-sodium diets are associated with a high prevalence of stroke and hypertension. Conversely, low-sodium diets are associated with a lower prevalence of hypertension. Clinical studies have shown that dietary sodium restriction lowers BP in many (but not all) patients with elevated BP. The exact mechanisms by which excess sodium leads to hypertension are not known.
Altered calcium homeostasis also may play an important role in the pathogenesis of hypertension. A lack of dietary calcium hypothetically can disturb the balance between intracellular and extracellular calcium, resulting in an increased intracellular calcium concentration. This imbalance can alter vascular smooth muscle function by increasing PVR. Some studies have shown that dietary calcium supplementation results in a modest BP reduction for patients with hypertension.
The role of potassium fluctuations is also inadequately understood. Potassium depletion may increase PVR, but the clinical significance of small serum potassium concentration changes is unclear. Furthermore, data demonstrating reduced CV risk with dietary potassium supplementation are very limited.
CLINICAL PRESENTATION
The clinical presentation of hypertension is described in Box 3-1.
BOX 3-1 CLINICAL PRESENTATION Hypertension
Diagnostic Considerations
![]() Hypertension is called the silent killer because most patients do not have symptoms. The primary physical finding is elevated BP. The diagnosis of hypertension cannot be made based on one elevated BP measurement. The average of two or more measurements taken during two or more clinical encounters is required to diagnose hypertension.1 This BP average should be used to establish a diagnosis, and then classify the stage of hypertension using Table 3-3.
Hypertension is called the silent killer because most patients do not have symptoms. The primary physical finding is elevated BP. The diagnosis of hypertension cannot be made based on one elevated BP measurement. The average of two or more measurements taken during two or more clinical encounters is required to diagnose hypertension.1 This BP average should be used to establish a diagnosis, and then classify the stage of hypertension using Table 3-3.
Measuring BP
The measurement of BP is a common routine medical screening tool that should be conducted at every healthcare encounter.1
Cuff Measurement Using Sphygmomanometry The most common procedure to measure BP in clinical practice is the indirect measurement of BP using sphygmomanometry.13 The appropriate procedure to indirectly measure BP using sphygmomanometry has been described by the AHA.13 It is imperative that the measurement equipment (inflation cuff, stethoscope, and manometer) meet certain national standards to ensure maximum quality and precision with measurement.
The AHA stepwise technique is recommended:
1. Patients should ideally refrain from nicotine and caffeine ingestion for 30 minutes and sit with lower back supported in a chair. Bare arm should be supported and resting near heart level. Feet should be flat on the floor (with legs not crossed). The measurement environment should be relatively quiet and provide privacy. Measuring BP in a position other than seated (supine or standing position) may be required under special circumstances (e.g., suspected orthostatic hypotension, or dehydration).
2. Measurement should begin only after a 5-minute period of rest.
3. A properly sized cuff (pediatric, small, regular, large, or extra large) should be used. The inflatable rubber bladder should be at least 80% and a width that is at least 40% of arm circumference.
4. The palpatory method should be used to estimate the SBP:
a. Place the cuff on the upper arm 2 to 3 cm above the antecubital fossa and attach it to the manometer (either a mercury or an aneroid).
b. Close the inflation valve and inflate the cuff to 70 mm Hg.
c. Palpate the radial pulse with the index and middle fingers of the opposite hand.
d. Inflate further in increments of 10 mm Hg until the radial pulse is no longer palpated.
e. Note the pressure at which the radial pulse is no longer palpated; this is the estimated SBP.
f. Release the pressure in the cuff by opening the valve.
5. The bell (not the diaphragm) of the stethoscope should be placed on the skin of the antecubital fossa, directly over where the brachial artery is palpated. The stethoscope earpieces should be inserted appropriately. The valve should be closed with the cuff, and then inflated to 30 mm Hg above the estimated SBP from the palpatory method. The valve should be slightly opened to slowly release pressure at a rate of 2 to 3 mm Hg/s.
6. The clinician should listen for Korotkoff sounds with the stethoscope. The first phase of Korotkoff sounds is the initial presence of clear tapping sounds. Note the pressure at the first recognition of these sounds. This is the SBP. As pressure deflates, note the pressure when all sounds disappear, right at the last sound. This is the DBP.
7. Measurements should be rounded to the nearest 2 mm Hg.
8. A second measurement should be obtained after at least 1 minute. If these values differ by more than 5 mm Hg, additional measurements should be obtained.
9. Neither the patient nor the observer should talk during measurement.
10. When first establishing care with a patient, BP should be measured in both arms. If consistent interarm differences exist, the arm with the higher value should be used.
Inaccuracies with indirect measurements result from inherent biologic variability of BP, inaccuracies related to suboptimal technique, and the white coat effect.13 Variations in BP occur with environmental temperature, the time of day and year, meals, physical activity, posture, alcohol, nicotine, and emotions. In the clinic setting, standard BP measurement procedures (e.g., appropriate rest period, correct technique, minimal number of measurements) are often not followed, which results in poor estimation of true BP. It is recommended that the stethoscope bell, rather than the diaphragm, be used for measurement, although some studies suggest little difference between two.13
Approximately 15% to 20% of patients have white coat hypertension, where BP values rise in a clinical setting but return to normal in nonclinical environments using home or ambulatory BP (ABP) measurements.13 Interestingly, the rise in BP dissipates gradually over several hours after leaving the clinical setting. It may or may not be precipitated by other stresses in the patient’s daily life. This is in contrast to masked hypertension, where a decrease in BP occurs in the clinical setting.14 With masked hypertension, home BP is hypertensive, while the in-office BP is normotensive or substantially lower than that at home. This situation may lead to undertreatment or lack of treatment for hypertension. Moreover, patients with either white coat or masked hypertension have a high risk of progressing to develop sustained hypertension, which can result in a higher risk of CV events compared with normotensive patients.15
Pseudohypertension is a falsely elevated BP measurement. It may be seen in the elderly, those with long-standing diabetes, or those with CKD due to rigid, calcified brachial arteries.13 In these patients, the true arterial BP when measured directly with intraarterial measurement (the most accurate measurement of BP) is much lower than that measured using the indirect cuff method. The Osler’s maneuver can be used to test for pseudohypertension. In this maneuver, the BP cuff is inflated above peak SBP. If the radial artery remains palpable, the patient has a positive Osler’s sign (rigid artery), which may indicate pseudohypertension.
Elderly patients with a wide pulse pressure may have an auscultatory gap that can lead to underestimated SBP or overestimated DBP measurements.13 In this situation, as the cuff pressure falls from the true SBP value, the Korotkoff sound may disappear (indicating a false DBP measurement), reappear (a false SBP measurement), and then disappear again at the true DBP value. When an auscultatory gap is present, Korotkoff sounds are usually heard when pressure in the cuff first starts to decrease after inflation. This may be eliminated by raising the arm overhead by 30 seconds before bringing it to the proper position and inflating the cuff. This maneuver decreases the intravascular volume and improves inflow thereby allowing Korotkoff sounds to be heard.13
Ambulatory and Self-BP Monitoring Ambulatory BP (ABP) monitoring using an automated device can document BP at frequent time intervals (e.g., every 15 to 30 minutes) throughout a 24-hour period.13 ABP values are usually lower than clinic-measured values. The definition of hypertension for ABP is ≥135/85 mm Hg during the day, ≥120/75 mm Hg nighttime (or asleep), and ≥130/80 mm Hg over 24 hours.13 For self BP monitoring, a BP ≥135/85 mm Hg is considered hypertensive. Self-BP measurements are collected by patients, preferably in the morning, using home monitoring devices.
Neither ABP nor self-BP monitoring is needed for the routine diagnosis of hypertension according to JNC7 guidelines; however, these modalities can enhance the ability to identify patients with white coat and masked hypertension.14 ABP and self-BP measurements may also be useful in evaluating and optimizing BP control for patients on antihypertensive drug therapy.14,16 ABP monitoring may be helpful for patients with apparent drug resistance, hypotensive symptoms while on antihypertensive therapy, episodic hypertension (e.g., white coat hypertension), autonomic dysfunction, and in identifying “nondippers” whose BP does not decrease by >10% during sleep and who may portend increased risk of BP-related complications.1,13
Limitations of ABP and self-BP measurements may prohibit routine use of such technology. These include complexity of use, costs, and lack of prospective outcome data describing normal ranges for these measurements. Although self-monitoring of BP at home is less complicated and less costly than ambulatory monitoring, patients may omit or fabricate readings, or have poor technique (e.g., not resting for adequate period of time, improper placement, wrong cuff size).
Clinical Evaluation
Frequently, the only sign of essential hypertension is elevated BP. The rest of the physical examination may be completely normal. However, a complete medical evaluation (a comprehensive medical history, physical examination, and laboratory and/or diagnostic tests) is recommended after diagnosis to (a) identify secondary causes, (b) identify other CV risk factors or comorbid conditions that may define prognosis and/or guide therapy, and (c) assess for the presence or absence of hypertension-associated target-organ damage.1 All patients with hypertension should have the tests described in Box 3-1 measured prior to initiating therapy.1 For patients without a history of coronary artery disease, noncoronary atherosclerotic vascular disease, left ventricular dysfunction, or diabetes, it is also important to estimate future risk of CV disease. The 10-year risk of fatal coronary heart disease or nonfatal MI can be estimated using Framingham risk scoring (http://www.nhlbi.nih.gov/guidelines/cholesterol/risk_tbl.htm).2
Secondary Causes
The most common secondary causes of hypertension are listed in Table 3-1. A complete medical evaluation should provide clues for identifying secondary hypertension.
Patients with secondary hypertension might have signs or symptoms suggestive of the underlying disorder. Patients with pheochromocytoma may have a history of paroxysmal headaches, sweating, tachycardia, and palpitations. Over half of these patients suffer from episodes of orthostatic hypotension. In primary hyperaldosteronism symptoms related to hypokalemia usually include muscle cramps and muscle weakness. Patients with Cushing’s syndrome may complain of weight gain, polyuria, edema, menstrual irregularities, recurrent acne, or muscular weakness and have several classic physical features (e.g., moon face, buffalo hump, hirsutism). Patients with coarctation of the aorta may have higher BP in the arms than in legs and diminished or even absent femoral pulses. Patients with renal artery stenosis may have an abdominal systolic–diastolic bruit.
Routine laboratory tests may also help identify secondary hypertension. Baseline hypokalemia may suggest mineralocorticoid-induced hypertension. Protein, red blood cells, and casts in the urine may indicate renovascular disease. Some laboratory tests are used specifically to diagnose secondary hypertension. These include plasma norepinephrine and urinary metanephrine for pheochromocytoma, plasma and urinary aldosterone concentrations for primary hyperaldosteronism, and plasma renin activity, captopril stimulation test, renal vein renin, and renal artery angiography for renovascular disease.
Certain drugs and other products can result in drug-induced hypertension (see Table 3-1). For some patients, the addition of these agents can be the cause of elevated BP or can exacerbate underlying hypertension. Identifying a temporal relationship between starting the suspected agent and developing elevated BP is most suggestive of drug-induced BP elevation.
Natural Course of Disease
Essential hypertension is usually preceded by elevated BP values that are in the prehypertension category. BP values may fluctuate between elevated and normal levels for an extended period of time. As the disease progresses, PVR increases, and BP elevation becomes chronic.
Hypertension-Associated Target-Organ Damage
Target-organ damage (see Box 3-1) can develop as a complication of hypertension. CV events (e.g., MI, cerebrovascular accidents, kidney failure) are clinical end points of hypertension-associated target-organ damage and are the primary causes of CV morbidity and mortality for patients with hypertension. The probability of CV events and CV morbidity and mortality in patients with hypertension is directly correlated with the severity of BP elevation.
Hypertension accelerates atherosclerosis and stimulates left ventricular and vascular dysfunction. These pathologic changes are thought to be secondary to both a chronic pressure overload and a variety of nonhemodynamic stimuli. Several nonhemodynamic disturbances have been implicated in these effects (e.g., the adrenergic system, RAAS, increased synthesis and secretion of endothelin I, and a decreased production of prostacyclin and nitric oxide). Atherosclerosis in hypertension is accompanied by proliferation of smooth muscle cells, lipid infiltration into the vascular endothelium, and enhancement of vascular calcium accumulation.
Cerebrovascular disease is a consequence of hypertension. A neurologic assessment can detect either gross neurologic deficits or a slight hemiparesis with some incoordination and hyperreflexia that are indicative of cerebrovascular disease. Stroke can result from lacunar infarcts caused by thrombotic occlusion of small vessels or intracerebral hemorrhage resulting from ruptured microaneurysms. Transient ischemic attacks secondary to atherosclerotic disease in the carotid arteries can also happen in patients with hypertension.
Retinopathies can occur in hypertension and may manifest as a variety of different findings. A funduscopic examination can detect hypertensive retinopathy, and the result can be categorized according to the Keith-Wagener-Barker retinopathy classification. Retinopathy manifests as arteriolar narrowing, focal arteriolar constrictions, arteriovenous crossing changes (nicking), retinal hemorrhages and exudates, and disk edema. Accelerated arteriosclerosis, a long-term consequence of essential hypertension, can cause nonspecific changes such as increased light reflex, increased tortuosity of vessels, and arteriovenous nicking. Focal arteriolar narrowing, retinal infarcts, and flame-shaped hemorrhages usually are suggestive of accelerated or malignant phase of hypertension. Papilledema is swelling of the optic disk caused by a breakdown in autoregulation of capillary blood flow in the presence of high pressure. It is usually only present in hypertensive emergencies.
Heart disease is the most well-identified form of target-organ damage. A thorough cardiac and pulmonary examination can identify cardiopulmonary abnormalities. Clinical manifestations include LVH, coronary heart disease (angina, prior MI, and prior coronary revascularization), and heart failure. These complications may lead to cardiac arrhythmias, angina, MI, and sudden death. Coronary disease (also called coronary heart disease) and associated CV events are the most common causes of death in patients with hypertension.
The kidney damage caused by hypertension is characterized pathologically by hyaline arteriosclerosis, hyperplastic arteriosclerosis, arteriolar hypertrophy, fibrinoid necrosis, and atheroma of the major renal arteries. Glomerular hyperfiltration and intraglomerular hypertension are early stages of hypertensive nephropathy. Albuminuria is followed by a gradual decline in renal function. The primary renal complication in hypertension is nephrosclerosis, which is secondary to arteriosclerosis. Atheromatous disease of a major renal artery may give rise to renal artery stenosis. Although overt kidney failure is an uncommon complication of essential hypertension, it is an important cause of end-stage kidney disease, especially in African Americans, Hispanics, and Native Americans.
The peripheral vasculature is a target organ. Physical examination of the vascular system can detect evidence of atherosclerosis, which may present as arterial bruits (aortic, abdominal, or peripheral), distended veins, diminished or absent peripheral arterial pulses, or lower extremity edema. PAD is a clinical condition that can result from atherosclerosis, which is accelerated in hypertension. Other CV risk factors (e.g., smoking) can increase the likelihood of PAD as well as all other forms of target-organ damage.
TREATMENT
Desired Outcomes
Overall Goal of Treatment
![]() The overall goal of treating hypertension is to reduce hypertension-associated morbidity and mortality.1 This morbidity and mortality is related to hypertension-associated target-organ damage (e.g., CV events, cerebrovascular events, heart failure, kidney disease). Reducing CV risk is the primary purpose of hypertension therapy and the specific choice of drug therapy should be determined by evidence demonstrating such CV risk reduction.
The overall goal of treating hypertension is to reduce hypertension-associated morbidity and mortality.1 This morbidity and mortality is related to hypertension-associated target-organ damage (e.g., CV events, cerebrovascular events, heart failure, kidney disease). Reducing CV risk is the primary purpose of hypertension therapy and the specific choice of drug therapy should be determined by evidence demonstrating such CV risk reduction.
Surrogate Targets—Blood Pressure Goals
![]() Treating patients with hypertension to achieve a desired target BP value is simply a surrogate goal of therapy. Reducing BP to goal does not guarantee prevention of hypertension-associated target-organ damage, but is associated with a lower risk of hypertension-associated target-organ damage.1 Targeting a goal BP value is a tool that clinicians use to evaluate response to therapy. It is the primary method used to determine the need for titration and regimen modification.
Treating patients with hypertension to achieve a desired target BP value is simply a surrogate goal of therapy. Reducing BP to goal does not guarantee prevention of hypertension-associated target-organ damage, but is associated with a lower risk of hypertension-associated target-organ damage.1 Targeting a goal BP value is a tool that clinicians use to evaluate response to therapy. It is the primary method used to determine the need for titration and regimen modification.
The JNC7 guidelines recommend BP goals for the management of hypertension (Box 3-2). A goal BP of <140/90 mm Hg is recommended for most patients for general prevention of CV events or CV disease (e.g., coronary artery disease).1 The JNC7, published in 2003, recommends a lower BP goal of <130/80 mm Hg for patients with diabetes or significant CKD. However, data supporting that this goal provides better reductions in CV events than a goal of <140/90 mm Hg are lacking. In 2013 the American Diabetes Association changed their recommended goal BP in patients with diabetes to <140/80 mm Hg.17 This was a significant change from their long-standing recommendation of <130/80 mm Hg. They do recommend that a SBP goal of <130 mm Hg may be appropriate for certain individuals (e.g., younger patients) if achieved without undue treatment burden, but the recommendation for most patients with diabetes is <140/80 mm Hg. Similarly, the Kidney Disease Improving Global Outcomes (KDIGO) guidelines recommend a BP goal of 140/90 mm Hg for patients with hypertension and CKD (nondialysis), with a lower BP goal of <130/80 mm Hg only for those patients who have persistent albuminuria (>30 mg urine albumin excretion per 24 hours or equivalent).18,19
BOX 3-2 Goal BP Values Recommended by the JNC7 in 2003, American Diabetes Association, and KDIGO
Until a new clinical guideline based on the NHLBI evidentiary review is published, clinicians should use the most recent recommended BP goals from these consensus statements of <140/90 mm Hg for most patients, <140/80 mm Hg for patients with diabetes, and <130/80 mm Hg for patients with persistent increased urinary albumin excretion (see Box 3-2).
Evidence Supporting <140/90 mm Hg in Most Patients Lower goal DBP values have been evaluated prospectively in the Hypertension Optimal Treatment (HOT) study.20 In this study, over 18,700 patients were randomized to target DBP values of ≤90, ≤85, or ≤80 mm Hg. Although the actual DBP values achieved were 85.2, 83.2, and 81.1 mm Hg, respectively, there were no significant differences in risk of major CV events when the three treatment groups were compared with each other among the total population. This lack of a benefit in reducing risk of CV events is consistent with findings from a 2009 Cochrane Collaboration systematic review that included seven clinical trials that evaluated different goal DBP values in hypertension.21 When the relationship between actual BP values and risk of CV events was evaluated, there was a trend that lower was better. The risk of major CV events was the lowest with a BP of 139/83 mm Hg, and lowest risk of stroke was with a BP of 142/80 mm Hg.
A major limitation of the HOT study and the 2009 Cochrane Collaboration review is the use of DBP goal values. SBP is more directly correlated to CV risk than DBP in most patients with hypertension, especially those above the age of 50. Therefore, data from the HOT study cannot answer this question. It is important to note that no J-curve relationship was seen. The J-curve hypothesis suggests that lowering BP too much might increase the risk of CV events.22 This theoretical hypothesis was described many years ago and was originally suggested in observational studies. Therefore, it remains an unproven hypothesis.
Limited data suggest lower is better when SBP goal values are targeted. The Cardio-Sys trial was a small open-label study in 1,111 patients with hypertension and without a history of diabetes.23 These patients had additional CV risk factors and roughly reflect a population with a Framingham risk score of 10% or greater. Patients were randomized to SBP goals of <140 mm Hg or <130 mm Hg. After a median of 2 years, the incidence of LVH was lower in the group randomized to a SBP goal of <130 mm Hg. Interestingly, the incidence of CV events, which was a secondary end point, was also significantly lower in the <130 mm Hg group. These data suggest that the optional lower BP goals may be better. However, LVH is only a surrogate end point for CV events, and the open-label nature of this study limits broad application to patient care.
Clinical Controversy…
Evidence Supporting Lower BP Goals in Diabetes A BP goal of <130/80 mm Hg was historically recommended for patients with diabetes for many years, by multiple organizations.1,2 The primary evidence supporting this recommendation was from the HOT study, where the only subgroup to show a lower risk of major CV events in the <80 mm Hg group versus the <90 mm Hg group was in patients with diabetes (n = 1,501).
The NHLBI-sponsored Action to Control Cardiovascular Risk in Diabetes Blood Pressure (ACCORD-BP) study questioned the benefit of lower BP goals for patients with diabetes.24 The ACCORD-BP was an open-label study that randomized 4,733 patients with type 2 diabetes to intensive therapy targeting a SBP of <120 mm Hg, or to standard therapy targeting a SBP <140 mm Hg for a mean followup of 4.7 years. After 1 year, an average of 3.4 medications was needed in the intensive therapy group to attain a mean SBP of 119.3 mm Hg, compared with an average of 2.1 medications in the standard therapy group to attain a mean SBP of 133.5 mm Hg. This difference was generally maintained throughout the study duration. However, there was no significant difference in the annual rate of the primary end point (nonfatal MI, nonfatal stroke, or CV death) between the two groups. The annual incidence of the secondary end point of stroke was lower with the intensive therapy group versus the standard therapy group, and this was the only prespecified end point that was different between the two groups.
Despite consensus guidelines historically recommending a BP goal of <130/80 mm Hg for patients with diabetes, evidence supporting this approach over a standard goal of <140/90 mm Hg is marginal, and comes at the cost of increased side effects (e.g., hypotension, hyperkalemia, bradycardia). While the ACCORD-BP provided additional evidence regarding BP goals for patients with diabetes, these data do not provide all of the clinical answers that are needed. The ACCORD-BP was open label, and those in the standard group (SBP <140 mm Hg) actually had SBP values that were closer to 130 mm Hg than to 140 mm Hg. Based on these data, in 2013 American Diabetes Association changed their recommendation to a goal BP of <140/80 mm Hg for most patients with hypertension and diabetes.17 The KDIGO guidelines recommend a BP goal of <140/90 mm Hg for patients with hypertension and CKD (non-dialysis) and a BP goal of <130/80 mm Hg only for those patients who have persistently increased urine albumin excretion.18
Avoiding Clinical Inertia
Although hypertension is one of the most common medical conditions, BP control rates are poor. Clinical inertia in hypertension has been defined as an office visit at which no therapeutic move was made to lower BP in a patient with uncontrolled hypertension.25 Clinical inertia is not the entire reason why many patients with hypertension do not achieve goal BP values. However, it is certainly a major reason that can be remedied simply through more aggressive treatment with drug therapy. This can involve initiating, titrating, or changing drug therapy.
General Approach to Treatment
Most patients should be placed on both lifestyle modifications and drug therapy concurrently after a diagnosis of hypertension is made. Lifestyle modification alone is appropriate for most patients with prehypertension. However, lifestyle modifications alone may not be adequate for patients with hypertension and either additional CV risk factors or hypertension-associated target-organ damage.
![]() The choice of initial drug therapy depends on the degree of BP elevation and presence of compelling indications (discussed in the Pharmacotherapy section below). Most patients with stage 1 hypertension should be initially treated with a first-line antihypertensive drug or the combination of two agents. Combination drug therapy is recommended for patients with more severe BP elevation (stage 2 hypertension), using preferably two first-line antihypertensive drugs. This general approach is outlined in Figure 3-2. There are six compelling indications where specific antihypertensive drug classes have evidence showing unique benefits in patients with hypertension and the listed compelling indication (Fig. 3-3).
The choice of initial drug therapy depends on the degree of BP elevation and presence of compelling indications (discussed in the Pharmacotherapy section below). Most patients with stage 1 hypertension should be initially treated with a first-line antihypertensive drug or the combination of two agents. Combination drug therapy is recommended for patients with more severe BP elevation (stage 2 hypertension), using preferably two first-line antihypertensive drugs. This general approach is outlined in Figure 3-2. There are six compelling indications where specific antihypertensive drug classes have evidence showing unique benefits in patients with hypertension and the listed compelling indication (Fig. 3-3).
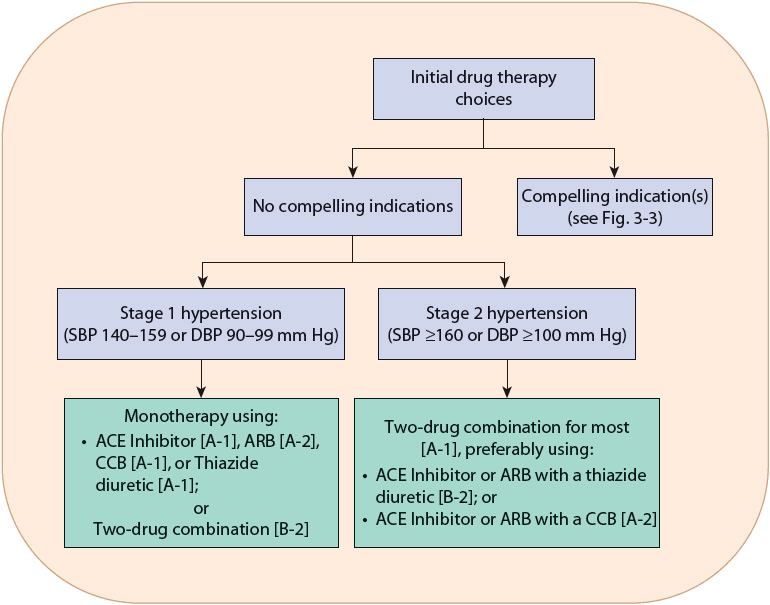
FIGURE 3-2 Algorithm for treatment of hypertension. Drug therapy recommendations are graded with strength of recommendation and quality of evidence in brackets. Strength of recommendations: A, B, and C are good, moderate, and poor evidence to support recommendation, respectively. Quality of evidence: (1) evidence from more than one properly randomized controlled trial; (2) evidence from at least one well-designed clinical trial with randomization, from cohort or case-controlled studies, or dramatic results from uncontrolled experiments or subgroup analyses; (3) evidence from opinions of respected authorities, based on clinical experience, descriptive studies, or reports of expert communities.
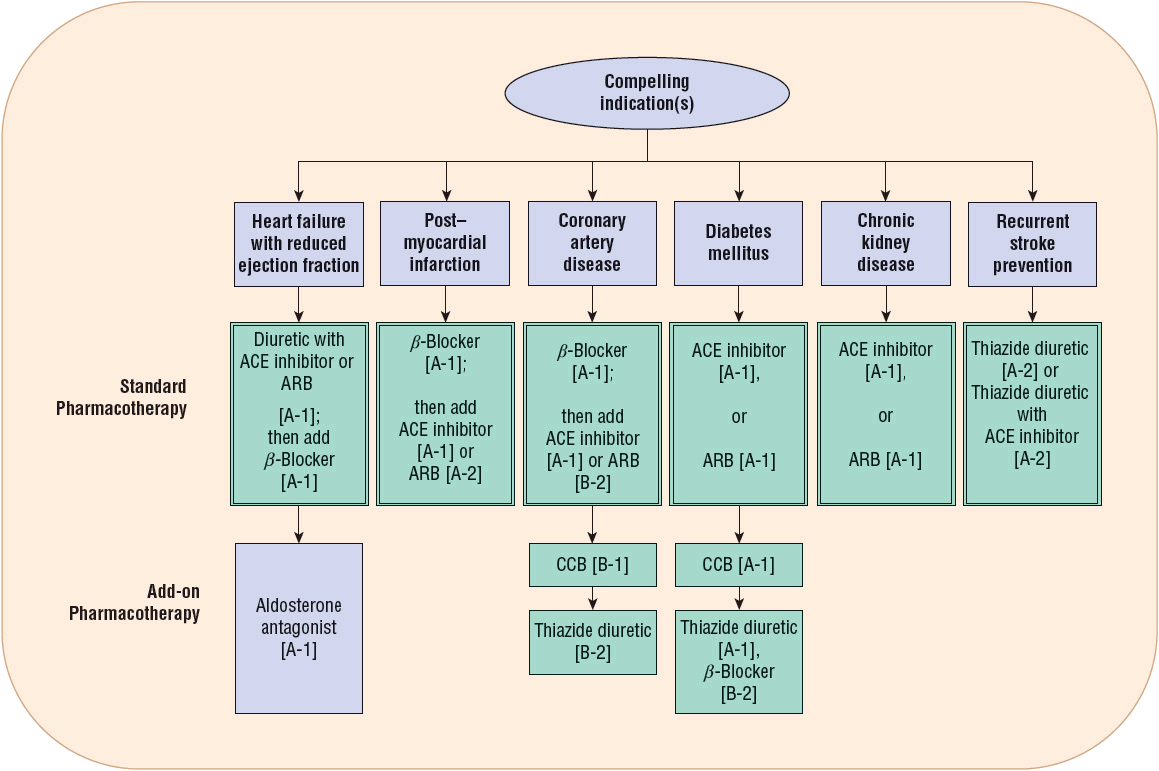
FIGURE 3-3 Compelling indications for individual drug classes. Compelling indications for specific drugs are evidenced-based recommendations from outcome studies or existing clinical guidelines. The order of drug therapies serves as a general guidance that should be balanced with clinical judgment and patient response; however, standard pharmacotherapy should be considered first-line recommendations, preferably in the order depicted. Then add-on pharmacotherapy recommendations are intended to further reduce risk of cardiovascular events when additional pharmacotherapy is needed to lower blood pressure to goal values. Blood pressure control should be managed concurrently with the compelling indication. Drug therapy recommendations are graded with strength of recommendation and quality of evidence in brackets. Strength of recommendations: A, B, and C are good, moderate, and poor evidence to support recommendation, respectively. Quality of evidence: (1) evidence from more than one properly randomized controlled trial; (2) evidence from at least one well-designed clinical trial with randomization, from cohort or case-controlled analytic studies or multiple time series, or dramatic results from uncontrolled experiments or subgroup analyses; (3) evidence from opinions of respected authorities, based on clinical experience, descriptive studies, or reports of expert communities.
Nonpharmacologic Therapy
![]() All patients with prehypertension and hypertension should be prescribed lifestyle modifications. Recommended modifications that have been shown to lower BP are listed in Table 3-4.1,26 They can provide small to moderate reductions in SBP. Aside from lowering BP in patients with known hypertension, lifestyle modification can decrease the progression to hypertension in patients with prehypertension BP values.26
All patients with prehypertension and hypertension should be prescribed lifestyle modifications. Recommended modifications that have been shown to lower BP are listed in Table 3-4.1,26 They can provide small to moderate reductions in SBP. Aside from lowering BP in patients with known hypertension, lifestyle modification can decrease the progression to hypertension in patients with prehypertension BP values.26
TABLE 3-4 Lifestyle Modifications to Prevent and Manage Hypertension

A sensible dietary program is one that is designed to reduce weight gradually (for overweight and obese patients) and one that restricts sodium intake with only moderate alcohol consumption. Successful implementation of dietary lifestyle modifications by clinicians requires aggressive promotion through reasonable patient education, encouragement, and continued reinforcement. Patients may better understand the rationale for dietary intervention in hypertension if they are provided the following three observations and facts26:
1. Weight loss, as little as 10 lb (4.5 kg), can decrease BP significantly in overweight patients.
2. Diets rich in fruits and vegetables and low in saturated fat have been shown to lower BP in patients with hypertension.
3. Most people experience some degree of BP reduction with sodium restriction.
The Dietary Approaches to Stop Hypertension (DASH) eating plan is a diet that is rich in fruits, vegetables, and low-fat dairy products with a reduced content of saturated and total fat. It is recommended as a reasonable and feasible diet that is proven to lower BP. Intake of sodium should be minimized as much as possible, ideally to 1.5 g/day, although an interim goal of <2.3 g/day may be reasonable considering the difficulty in achieving these low intakes. Patients should be aware of the multiple sources of dietary sodium (e.g., processed foods, soups, table salt) so that they may follow these recommendations. Potassium intake should be encouraged through fruits and vegetables with high content (ideally 4.7 g/day) in those with normal kidney function or without impaired potassium excretion. Excessive alcohol use can either cause or worsen hypertension. Patients with hypertension who drink alcoholic beverages should restrict their daily intake.
Carefully designed programs of physical activity can lower BP. Regular physical activity for at least 30 minutes most days of the week is recommended for all adults, with at least 60 minutes recommended for adults attempting to lose weight or maintain weight loss.27 Studies have shown that aerobic exercise can reduce BP, even in the absence of weight loss. Patients should consult their physicians before starting an exercise program, especially those with CV and/or hypertension-associated target-organ disease.
Cigarette smoking is not a secondary cause of essential hypertension; it is a major, independent, modifiable risk factor for CV disease. Patients with hypertension who smoke should be counseled regarding the additional health risks that result from smoking. Moreover, the potential benefits that cessation can provide should be explained to encourage cessation.
Pharmacotherapy
![]() ACE inhibitors, angiotensin II receptor blockers (ARBs), calcium channel blockers (CCBs), and thiazide diuretics are considered primary antihypertensive agents that may be acceptable first-line options (Table 3-5). These agents should be used to treat the majority of patients with hypertension because evidence from outcome data have demonstrated CV risk reduction benefits with these classes. Several have subclasses where significant differences in mechanism of action, clinical use, side effects, or evidence from outcome studies exist. β-Blockers are effective antihypertensive agents that previously were considered primary agents. They are now preferred either to treat a specific compelling indication or in combination with one or more of the aforementioned primary antihypertensive agents for patients without a compelling indication. Other antihypertensive drug classes are considered alternative drug classes that may be used in select patients after first-line agents (Table 3-6).
ACE inhibitors, angiotensin II receptor blockers (ARBs), calcium channel blockers (CCBs), and thiazide diuretics are considered primary antihypertensive agents that may be acceptable first-line options (Table 3-5). These agents should be used to treat the majority of patients with hypertension because evidence from outcome data have demonstrated CV risk reduction benefits with these classes. Several have subclasses where significant differences in mechanism of action, clinical use, side effects, or evidence from outcome studies exist. β-Blockers are effective antihypertensive agents that previously were considered primary agents. They are now preferred either to treat a specific compelling indication or in combination with one or more of the aforementioned primary antihypertensive agents for patients without a compelling indication. Other antihypertensive drug classes are considered alternative drug classes that may be used in select patients after first-line agents (Table 3-6).
TABLE 3-5 First-Line and Other Common Antihypertensive Agents
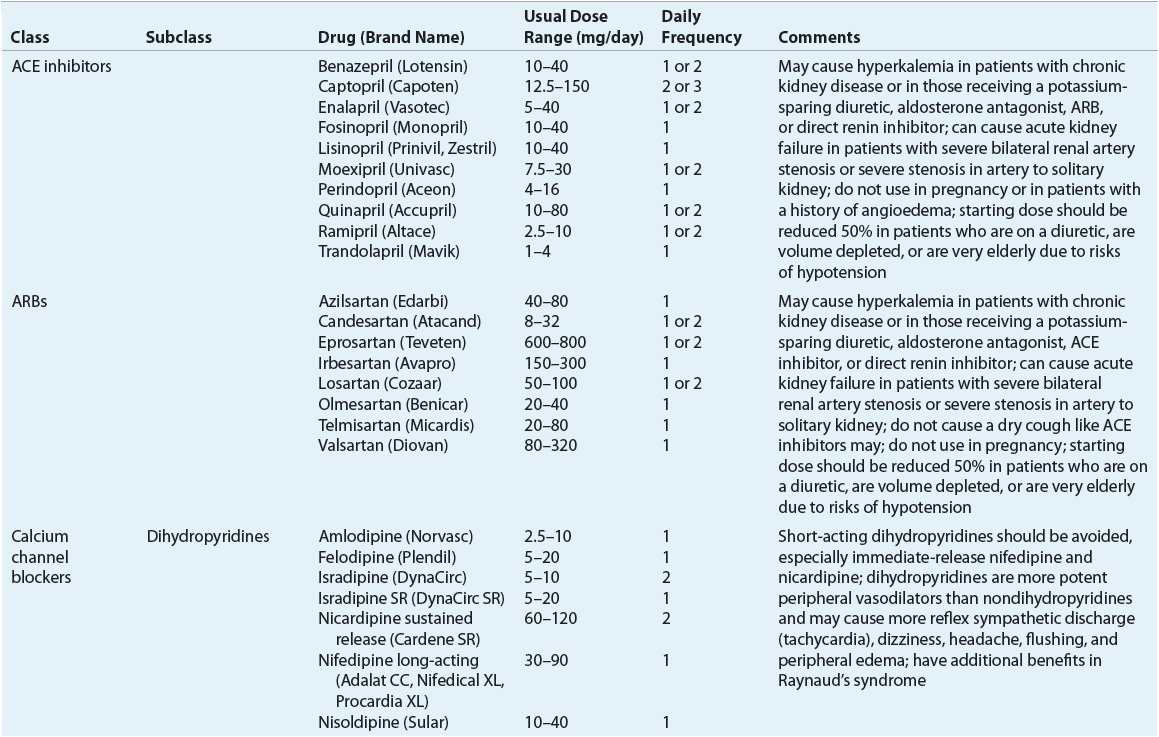
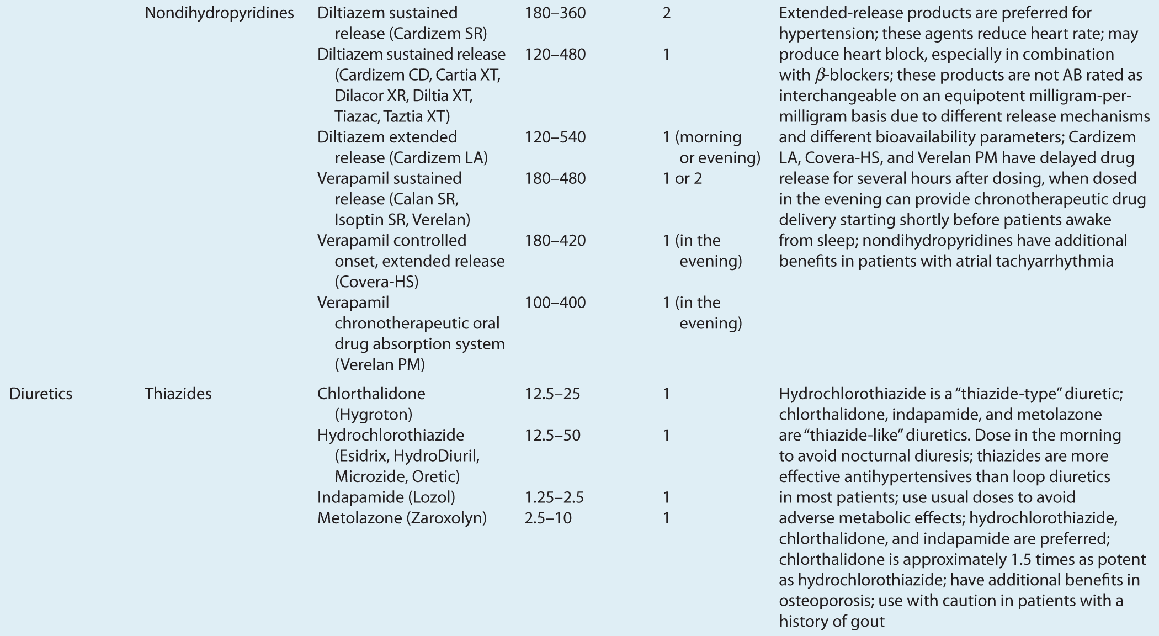
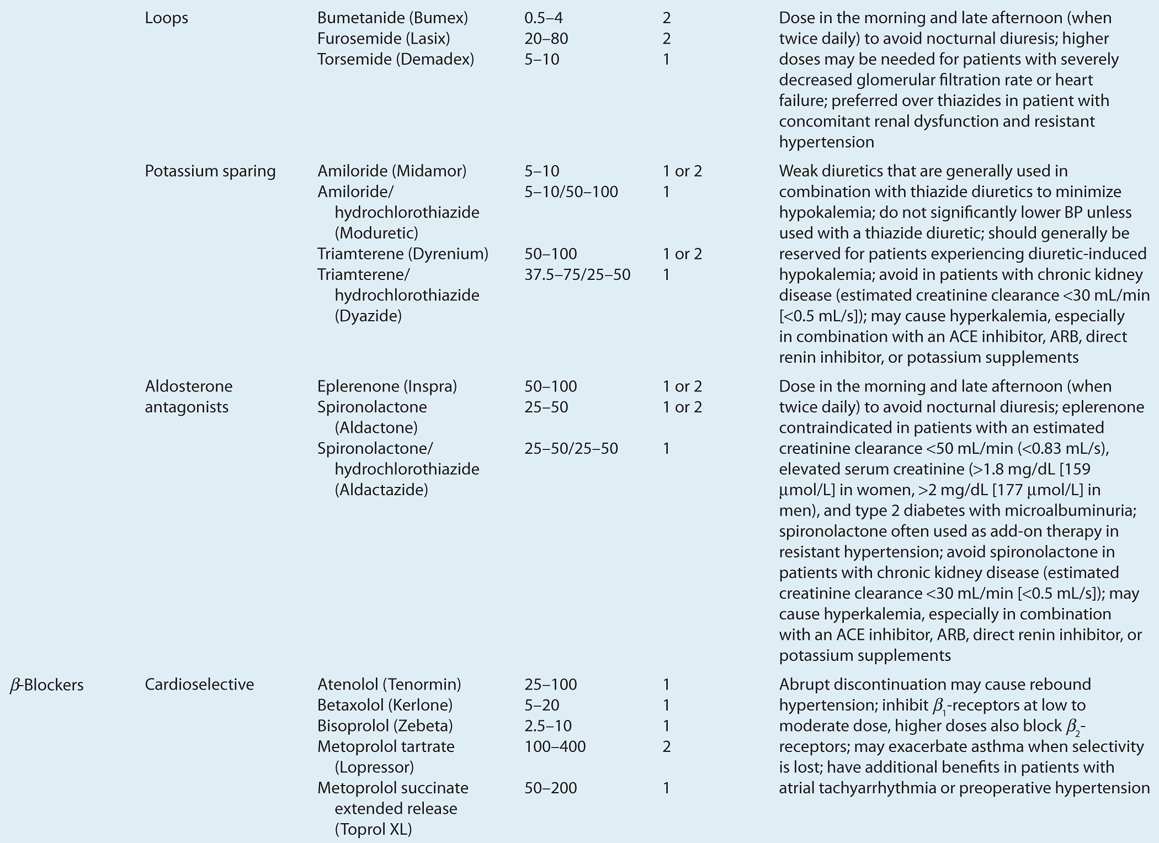
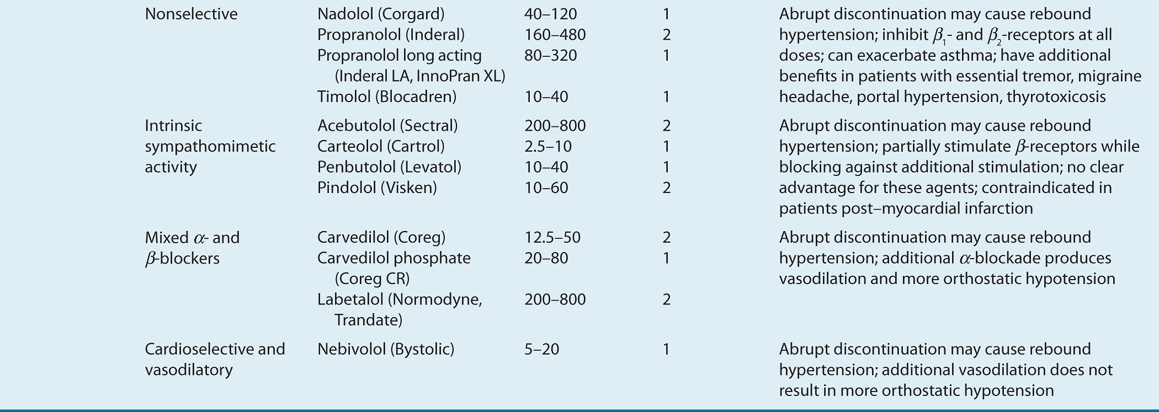
TABLE 3-6 Alternative Antihypertensive Agents
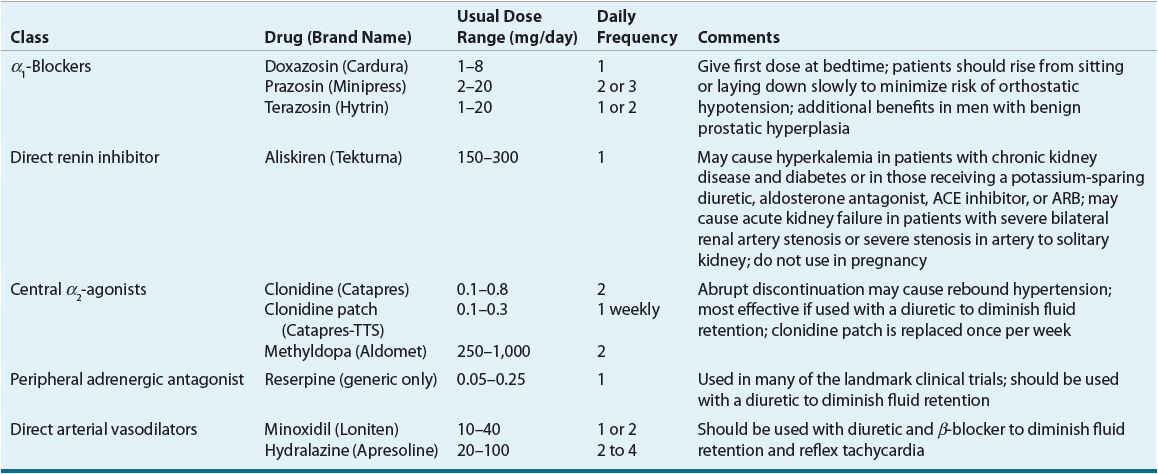
Thiazide Diuretics as Historical First-Line Agents
Landmark placebo-controlled clinical trials demonstrate that thiazide diuretic therapy irrefutably reduces risk of CV morbidity and mortality.28 The Systolic Hypertension in the Elderly Program (SHEP),8 Swedish Trial in Old Patients with Hypertension (STOP-Hypertension),9 and Medical Research Council (MRC)10 studies showed significant reductions in stroke, MI, all-cause CV disease, and mortality with thiazide diuretic–based therapy versus placebo. These trials allowed for β-blockers as add-on therapy for BP control. Newer agents (e.g., ACE inhibitors, ARBs, and CCBs) were not available at the time of these studies. However, subsequent clinical trials have compared these newer antihypertensive agents with thiazide diuretics.29–34 These data show similar effects, but most trials used a prospective open-label, blinded end point (PROBE) study methodology that is not double-blinded and limited their ability to prove equivalence of newer drugs to diuretics. Other prospective trials have compared different primary antihypertensive agents with each other.35,36 Although these studies used head-to-head comparisons, they did not use a thiazide diuretic as their comparator treatment.
The Antihypertensive and Lipid Lowering Treatment to Prevent Heart Attack Trial (ALLHAT) The results of the ALLHAT were the deciding evidence that the JNC7 used to justify thiazide diuretics as first-line therapy.32 It was designed to test the hypothesis that newer antihypertensive agents (an α-blocker, ACE inhibitor, or dihydropyridine CCB) would be superior to thiazide diuretic–based therapy. The primary objective was to compare the combined end point of fatal CHD and nonfatal MI. Other hypertension-related complications (e.g., heart failure, stroke) were evaluated as secondary end points. This was the largest prospective hypertension trial ever conducted and included 42,418 patients aged 55 and older with hypertension and one additional CV risk factor. This double-blind trial randomized patients to chlorthalidone-, amlodipine-, doxazosin-, or lisinopril-based therapy for a mean of 4.9 years.
The doxazosin arm was terminated early when a significantly higher risk of heart failure versus chlorthalidone was observed.37 The other arms were continued as scheduled and no significant differences in the primary end point was seen between the chlorthalidone and lisinopril or amlodipine treatment groups. However, chlorthalidone had statistically fewer secondary end points than amlodipine (heart failure) and lisinopril (combined CV disease, heart failure, and stroke). The study conclusions were that chlorthalidone-based therapy was superior in preventing one or more major forms of CV disease and was less expensive than amlodipine- or lisinopril-based therapy.
ALLHAT was designed as a superiority study with the hypothesis that amlodipine, doxazosin, and lisinopril would be better than chlorthalidone.38 It did not prove this hypothesis because the primary end point was no different between chlorthalidone, amlodipine, and lisinopril. Many subgroup analyses of specific populations (e.g., black patients, CKD, diabetes) from the ALLHAT have been conducted to assess response in certain unique patient populations.39–41 Surprisingly, none of these analyses demonstrated superior CV event reductions with lisinopril or amlodipine versus chlorthalidone. Overall, thiazide diuretics remain unsurpassed in their ability to reduce CV morbidity and mortality in most patients.
JNC7 guidelines (from 2003) recommend thiazide diuretics as a first-line therapy option for most patients, and are consistent with the historical treatment of hypertension.1 The AHA 2007 guidelines clearly identify thiazide diuretics as a first-line therapy option, comparable to an ACE inhibitor, ARB, or CCB for first-line therapy. Contrary to the historical preference to use a thiazide diuretic as the preferred first-line agent for treating most patients with hypertension, they are simply one of four first-line drug therapy options. Figure 3-2 displays the algorithm for the treatment of hypertension and highlights that four drug classes are considered first-line agents for patients without a compelling indication for a specific drug class.
Clinical Controversy…
ACE Inhibitors, ARBs, and CCBs as First-Line Agents
Clinical trial data cumulatively demonstrate that ACE inhibitor–, CCB-, or ARB-based antihypertensive therapy reduces CV events. These agents may be used for patients without compelling indications as a first-line therapy. The Blood Pressure Lowering Treatment Trialists’ Collaboration has evaluated the incidence of major CV events and death among different antihypertensive drug classes from 29 major randomized trials in 162,341 patients.42 In placebo-controlled trials, the incidences of major CV events were significantly lower with ACE inhibitor– and CCB-based regimens versus placebo. Although there were differences in the incidences of certain CV events in some comparisons (e.g., stroke was lower with diuretic or CCB-based regiments versus ACE inhibitor–based regimens), there were no differences in total major CV events when ACE inhibitors, CCBs, or diuretics were compared with each other. In studies evaluating ARB-based therapy to control regimens, the incidence of major CV events was lower with ARB-based therapy. However, the control regimens used in these comparisons included both active antihypertensive drug therapies and placebo.
Data from meta-analyses may not be as influential as data from well-designed, prospective, randomized controlled trials (e.g., the ALLHAT). However, they provide clinically useful data that support using ACE inhibitor–, CCB-, or ARB-based treatment for hypertension as first-line antihypertensive agents. Clinicians can use meta-analyses data as supporting evidence when selecting a first-line antihypertensive regimen for hypertension in most patients.
Other major consensus guidelines recommend multiple first-line options for treating hypertension in most patients. The 2013 European Society of Hypertension/European Society of Cardiology guidelines and the 2011 UK’s National Institute for Health and the Clinical Excellence guidelines list more than one drug therapy option as an acceptable first-line treatment approach.43,44 The European Society of Hypertension/European Society of Cardiology guidelines are founded on the principle that CV risk reduction is a function of BP control that is largely independent of specific antihypertensives.43 The UK guidelines stratify patients based on age and race; they recommend an ACE inhibitor or ARB first line for patients under the age of 55, and a CCB first line for patients age 55 or older or for black patients.44
![]() β-Blockers Versus First-Line Agents Clinical trial data cumulatively suggest that β-blockers may not reduce CV events to the extent that ACE inhibitors, ARBs, CCBs, or thiazide diuretics do. These data are from three meta-analyses of clinical trials evaluating β-blocker–based therapy for hypertension.45–48 Overall, these analyses demonstrated fewer reductions in CV events with β-blocker–based antihypertensive therapy compared mostly with ACE inhibitor– and CCB-based therapy. Although comparative data with ARB-based therapy are more limited, a similar trend was observed.
β-Blockers Versus First-Line Agents Clinical trial data cumulatively suggest that β-blockers may not reduce CV events to the extent that ACE inhibitors, ARBs, CCBs, or thiazide diuretics do. These data are from three meta-analyses of clinical trials evaluating β-blocker–based therapy for hypertension.45–48 Overall, these analyses demonstrated fewer reductions in CV events with β-blocker–based antihypertensive therapy compared mostly with ACE inhibitor– and CCB-based therapy. Although comparative data with ARB-based therapy are more limited, a similar trend was observed.
Meta-analyses data evaluating β-blockers and their ability to reduce CV events have limitations. Most studies that were included used atenolol as the β-blocker studied. Therefore, it is possible that atenolol is the only β-blocker that reduces CV events less than the other primary antihypertensive drug classes. However, consensus guidelines do extrapolate these findings to the β-blocker drug class in general.2 In the absence of a compelling indication, the 2011 UK guidelines recommend a β-blocker as fourth-line therapy, only after other primary antihypertensive agents (e.g., ACE inhibitor or ARB, CCB, thiazide diuretic) have been used.44 These findings also call in question the validity of results from prominent prospective, controlled clinical trials evaluating antihypertensive drug therapy that use β-blocker–based therapy, especially atenolol, as the primary comparator.31,36 Of note, these studies used once-daily atenolol, which may be inadequate based on the shorter half-life of this agent.
β-Blocker therapy for patients without compelling indications still has a role in the management of hypertension. It is important for clinicians to remember that β-blocker–based antihypertensive therapy does not increase risk of CV events; β-blocker–based therapy reduces risk of CV events compared with no antihypertensive therapy. Using a β-blocker as a primary antihypertensive agent is optimal when an ACE inhibitor, ARB, CCB, or thiazide diuretic cannot be used as the primary agent. β-Blockers still have an important add-on role after first-line agents to reduce BP in patients with hypertension but without compelling indications.



