Human Immunodeficiency Viruses: Replication
Eric O. Freed
Malcolm A. Martin
Introduction
In the late 1970s and early 1980s, previously healthy patients with symptoms of immunologic dysfunction sought advice and treatment from physicians in the United States and Europe. This new and unusual syndrome was characterized by generalized lymphadenopathy, opportunistic infections (typically Pneumocystis carinii pneumonia, cytomegalovirus-associated retinitis, and cryptococcal meningitis), and a variety of unusual cancers (non-Hodgkin’s lymphoma and Kaposi’s sarcoma). A common accompanying laboratory finding in affected individuals was marked depletion of the CD4+ T-lymphocyte subset in the peripheral blood. The disease was first brought to the attention of the general medical community in June 1981 when the Centers for Disease Control and Prevention described five California men with severe immunodeficiency in the Morbidity and Mortality Weekly Report529 (Fig. 49.1). This notification was followed by several reports describing male homosexuals and intravenous drug users with impaired immune systems characterized by T lymphocytes that responded poorly to antigen and mitogen stimulation in functional assays. Within several months, it became clear that a similar immunodeficiency syndrome was also affecting other groups, including hemophiliacs, blood transfusion recipients, recent Haitian immigrants, and, most significantly, sexual partners or children of members of the various risk groups.
The emerging epidemiologic pattern suggested that the new disease was transmitted by a novel pathogen in contaminated blood or following sexual intercourse with an affected individual. Between late 1981 and early 1983, a variety of microorganisms were proposed as possible causes of acquired immunodeficiency syndrome, or AIDS, as the disease was soon called. In the spring of 1983, Montagnier and colleagues32 at the Pasteur Institute reported the isolation of an agent from the lymph nodes of an asymptomatic individual who presented with generalized lymphadenopathy of unknown origin. During its replication in cultured cells, the lymphadenopathy-associated virus, or LAV as it was named, released high titers of progeny virions that contained magnesium-dependent reverse transcriptase (RT) activity and exhibited electron microscopic (EM) features typical of retroviruses. However, unlike the commonly studied retroviruses such as the avian leukosis viruses (ALVs) and the murine and feline leukemia viruses (MLVs and FeLVs), LAV was highly cytopathic in human peripheral blood mononuclear cells (PBMCs), specifically killing CD4+ T lymphocytes in cell cultures.532 Gallo and colleagues608,658 at the National Institutes of Health subsequently reported the isolation of a retrovirus from an AIDS patient, which they named human T-cell leukemia virus type III (HTLV-III) to distinguish it from the noncytopathic HTLV-1, and obtained the first serologic evidence linking exposure to LAV-like retroviruses and immunodeficient individuals from the various groups at risk. The new retrovirus, associated with AIDS in the United States, Europe, and central Africa and exhibiting morphologic and genetic characteristics typical of the lentivirus genus, was named human immunodeficiency virus, or HIV140 (and subsequently HIV-1). In 1986, a related but immunologically distinct human retrovirus (now called HIV-2) was recovered from individuals residing in several West African countries such as Senegal, the Ivory Coast, and Guinea-Bissau.139 Prior to the discovery and use of highly active
antiretroviral therapy (HAART), the risk of death for HIV-1–infected persons was 40-fold higher than the mortality in the general population, whereas HIV-2–infected individuals had a 2- to 5-fold higher risk.57,293,614
antiretroviral therapy (HAART), the risk of death for HIV-1–infected persons was 40-fold higher than the mortality in the general population, whereas HIV-2–infected individuals had a 2- to 5-fold higher risk.57,293,614
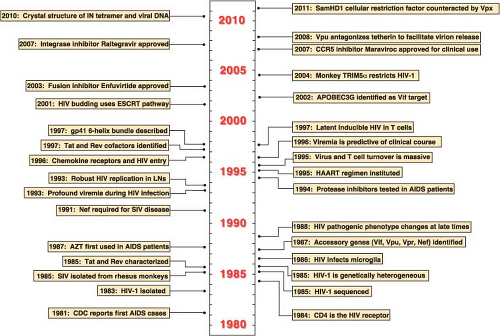 Figure 49.1. Landmarks in human immunodeficiency virus (HIV) research. Important discoveries and technical or conceptual advances are noted on either side of the central time line. |
Classification of Human Immunodeficiency Viruses
One of the first features the Pasteur Institute group noted about HIV-1 was its particle-associated RT activity, a property that placed the new agent in the Retroviridae family. This feature was consistent with EM analyses of particles released from infected cell cultures, which revealed 100- to 120-nm enveloped virions, similar in size and morphology to previously studied retroviruses. The mature HIV-1 particles contained a cone-shaped cylindrical core reminiscent of that previously described for visna virus (Fig. 49.2A).269 The cloning and sequencing of proviral DNA, initially purified from productively infected cultures of PBMC/T-cell leukemia lines (T-cell lines), indicated that HIV-1 not only possessed a genomic organization related to other replication-competent retroviruses but also was placed taxonomically in the Lentivirus genus.620,786 This relationship is shown diagrammatically in Figure 47.2 of Chapter 47. As their name suggests, lentiviruses were known to cause slow, unremitting disease in sheep, goats, and horses and to target various lineages of hematopoietic cells, particularly monocytes/macrophages and lymphocytes.
After the isolation, molecular cloning, and initial classification of HIV-1, several genetically distinct primate lentiviruses were subsequently discovered and their phylogenetic relationships to HIV-1 were determined. For example, viruses isolated from captive macaques or feral monkey species in Africa were shown to possess particle morphologies and genomic organizations similar to those of HIV-1 (described in Chapter 51). Because inoculation of Asian macaque species, such as rhesus monkeys, with these newly discovered agents recovered from African monkeys induced an AIDS-like illness,159 these viruses were named simian immunodeficiency virus (SIV) to distinguish them from the human viruses, HIV-1 and HIV-2. The phylogenetic relationships of the two human lentiviruses are depicted in Figure 49.3; the detailed genetic interrelationships of all members of the primate lentivirus genus are presented in Figure 52.1B of Chapter 52. HIV-2 is more closely related to SIVsm,320 a virus indigenous to African sooty mangabey monkeys, than to HIV-1. Because of the close contact between
humans and monkeys, which are hunted for food or kept as pets in West Africa, it is currently thought that HIV-2 represents a zoonotic transmission of SIVsm from monkeys to humans.253
humans and monkeys, which are hunted for food or kept as pets in West Africa, it is currently thought that HIV-2 represents a zoonotic transmission of SIVsm from monkeys to humans.253
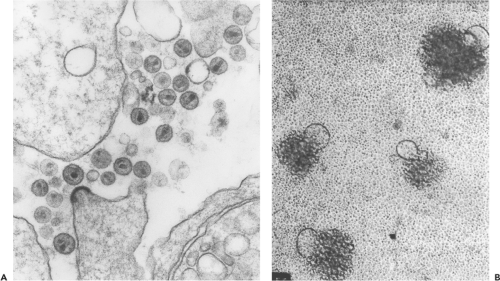 Figure 49.2. Primate lentiviruses have a distinct morphology and can induce syncytia during productive infections. A: Electron micrograph showing a single human immunodeficiency virus 1 (HIV-1) particle in the process of budding from infected cultured human peripheral blood mononuclear cells (PBMCs) and several mature virions containing the characteristic conical or bullet-shaped core (×100,000). (A courtesy of Dr. Jan Orenstein.) B: Typical ballooning syncytia induced by primate lentiviruses as visualized by inverted light microscopy at the time of peak virus production. |
Based on previously studied replication-competent retroviruses (e.g., MLV, ALV, and HTLV), it was originally thought that HIV-1 would be genetically homogeneous. However, as proviral DNAs corresponding to HIV-1 isolates from Europe, North America, and Africa, became available and were compared to one another, their extensive genetic heterogeneity became apparent.44 No two HIV-1 isolates were identical. When subjected to nucleotide sequence analysis, even HIV-1 samples recovered from a single individual exhibited significant heterology. Although nucleotide changes were distributed throughout the HIV-1 genome, the greatest variability occurred in the gene encoding the envelope (Env) glycoprotein, gp160. The term quasispecies was subsequently coined to describe the pool of diverse and changing populations of virus present in an HIV-1–infected individual.521 Nonetheless, a highly conserved immunodominant domain, located within gp41 Env-coding sequences, was found to elicit antibodies in exposed individuals, reactive with HIV-1 samples of diverse geographic origin.263 Although not neutralizing, these antibodies could be used in enzyme-linked immunosorbent assay (ELISA) and immunoblotting assays to identify virus-infected persons.
 Figure 49.3. Phylogenetic relationships of human immunodeficiency virus 1 (HIV-1) and HIV-2 based on identity of pol gene sequences. Simian immunodeficiency virus (SIV)cpz and SIVsm are nonhuman primate lentiviruses recovered from a chimpanzee and sooty mangabey monkey, respectively. |
Several factors contribute to the extraordinary genetic heterogeneity of HIV-1: (a) error-prone viral DNA synthesis during reverse transcription (1 to 2 × 10−5 mutations/nucleotide/replication cycle in tissue culture systems; 5 × 10−5 within a patient), (b) high recombination frequencies accompanying reverse transcription, (c) high levels of progeny virus production in vivo (109 particles/day; millions of replication cycles/year), and (d) large numbers of infected individuals.141,359,491,592,759 It has been estimated that within an HIV-1–infected person, viral genetic diversity increases by 1% per year from the founder virus strain during the early asymptomatic phase of the infection.689
The earliest phylogenetic analyses of HIV-1 isolates focused on samples from Europe, North America, and Africa;
discrete clusters of viruses were identified from these geographical regions. Distinct genetic subtypes or clades of HIV-1 were subsequently defined and classified into four groups: M (main), O (outlier), N (non-M, non-O), and P (Fig. 49.4A). The M group of HIV-1, which includes more than 98% of the global virus isolates, consists of nine clades or subgroups (A, B, C, D, F, G, H, J, and K) and 49 circulating recombinant forms (CRFs).462,511,588,632 HIV-1 group O isolates have been recovered from individuals living in Cameroon, Gabon, and Equatorial Guinea; their genomes share approximately 65% identity with group M viruses.742 Early in the epidemic, group O strains may have been responsible for more than 20% of HIV-1 infections in Cameroon but are presently associated with approximately only 1%.773 Group N HIV-1 strains have been identified in 13 infected Cameroonians and are detectable using currently available immunoassays. Very recently, HIV-1 isolates, identified in two Cameroonians, have been classified as members of the new HIV-1 group P.764
discrete clusters of viruses were identified from these geographical regions. Distinct genetic subtypes or clades of HIV-1 were subsequently defined and classified into four groups: M (main), O (outlier), N (non-M, non-O), and P (Fig. 49.4A). The M group of HIV-1, which includes more than 98% of the global virus isolates, consists of nine clades or subgroups (A, B, C, D, F, G, H, J, and K) and 49 circulating recombinant forms (CRFs).462,511,588,632 HIV-1 group O isolates have been recovered from individuals living in Cameroon, Gabon, and Equatorial Guinea; their genomes share approximately 65% identity with group M viruses.742 Early in the epidemic, group O strains may have been responsible for more than 20% of HIV-1 infections in Cameroon but are presently associated with approximately only 1%.773 Group N HIV-1 strains have been identified in 13 infected Cameroonians and are detectable using currently available immunoassays. Very recently, HIV-1 isolates, identified in two Cameroonians, have been classified as members of the new HIV-1 group P.764
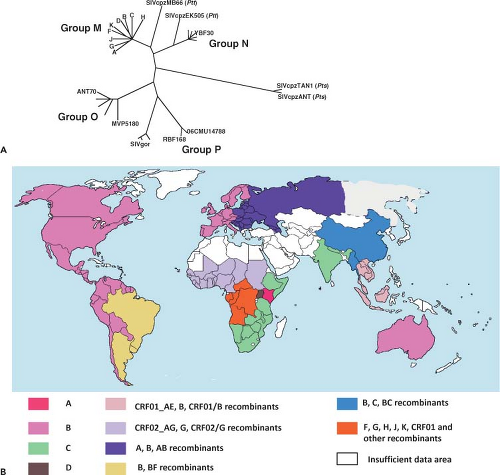 Figure 49.4. Human immunodeficiency virus 1 (HIV-1) genetic subtypes and their worldwide distribution. A: Phylogenetic relationships of HIV-1 groups M, N, O, and P with different simian immunodeficiency virus (SIVcpz and SIVgpr) isolates. This maximum-likelihood tree is adapted from Foster JL, Denial SJ, Temple BR, et al. Mechanisms of HIV-1 Nef function and intracellular signaling. J Neuroimmune Pharmacol 2011;6:230–246 with permission. B: The global prevalence of HIV-1 subgroups with the predominant clades or circulating recombinant forms (CRFs) in each geographic region indicated. (Courtesy Dr. Sodsai Tovanabutra, U.S. Military HIV Research Program, Henry Jackson Foundation.) |
As shown in Figure 49.4A, the nine clades comprising group M HIV-1 isolates are phylogenetically equidistant from one another, with internucleotide distances ranging from 15% to 20% in the gag gene and 20% to 30% in env.632 Within-clade distances range from 3% to 10% for gag and 5% to 12% for env; up to 10% sequence diversity in the env gene has been reported for a single infected person.412 All of the HIV-1 group M subtypes can be found in Africa (Fig. 49.4B). With the rapid spread of HIV-1 to southern Africa during the mid- to late 1990s, clade C viruses have become the dominant subtype and now account for nearly half of HIV-1 infections worldwide.577 Clade A viruses, the most common HIV-1 subtype in Africa early in the epidemic, currently represent about 12% of infections globally, and are present mainly in Eastern and Central African countries. Clade B viruses, the most intensively studied HIV-1 subtype, remain the most prevalent isolates in Europe, North America, and Australia.
High rates of genetic recombination, a hallmark of retroviruses, are generated as a consequence of a single cell being infected by genetically distinct viral strains and arise as a result of template switching between two different viral RNAs during the reverse transcription reaction (see section on Reverse Transcription). By 1995, it became apparent that a significant fraction of the global diversity of HIV-1 group M viruses included interclade CRFs, which arose by the intermixing of viruses co-circulating in a particular geographical locale. It is now appreciated that HIV-1 recombinants will be found in geographical areas such as Africa, South America, and Southeast Asia where multiple HIV-1 subtypes coexist and account for nearly 20% of circulating HIV-1 strains globally. Molecularly, the genomes of these CRFs resemble patchwork mosaics, with juxtaposed diverse HIV-1 subtype segments, reflecting the multiple crossover events contributing to their generation. Most HIV-1 recombinants have arisen in Africa and a majority contains segments originally derived from clade A viruses. Although an intergroup (groups M and O) recombinant virus has been described,588 no recombination between HIV-1 and HIV-2 has been reported, although mixed in vivo infections involving the latter are known to occur.279
A primate lentivirus designated SIVcpz, with a genomic structure very similar to that of HIV-1, including its signature vpu gene, has been isolated from two chimpanzee subspecies, Pan troglodytes troglodytes (ptt) and Pan troglodytes schweinfurthii (pts). The prevalence of SIVcpz recovered from ptt animals (SIVcpzPtt) in the wild varies widely (0 to 35%) compared to the more even and higher distribution of SIVs infecting sooty mangabeys and African green monkeys. Phylogenetically, the SIVcpzPtt strains are related to HIV-1 groups M and N but not to group O or SIVcpzPts (Fig. 49.4A), and geographically separated chimpanzee populations carry distinct genetic lineages of SIVcpzPtt.385 A prospective study of habituated chimpanzees in Tanzania, using noninvasive approaches to acquire fecal and urine specimens for antiviral antibody and nucleic acid assays, reported that SIVcpz-infected animals experienced a 10- to 16-fold increased risk of death and virus-positive females had decreased fertility and lower infant survival than uninfected chimpanzees, indicating that these infections are pathogenic in the wild.384 It has been suggested that SIVcpzPtt is the source of recently described SIVgor lineages recovered from Western lowland gorillas.741
It is now generally accepted that cross-species transmission to humans of SIVcpz in Central Africa and SIVsm in West Africa gave rise to HIV-1 and HIV-2, respectively.252,253 Based on nucleotide substitutions over time, a common ancestor of HIV-1 group M has been proposed to have emerged in the 1920s,813 whereas progenitors for HIV-2 groups A and B have been dated to the 1940s.434 The SIVcpz strains giving rise to HIV-1 have been introduced into humans at least four times (as reflected in groups M, N, O, and P). It is thought that the source of HIV-1 group M, the cause of the worldwide AIDS epidemic, originated in ptt chimpanzees living in southeast Cameroon.385 Infected humans very likely migrated from there, via tributaries of the Congo River, to Kinshasa, Republic of the Congo, where HIV-1 transmission greatly accelerated. Of the numerous species-specific strains of SIV that have been identified in more than 40 different species of African monkeys (see Chapter 51), none are related to SIVcpz across the entire viral genome. However, Vpu-coding sequences, unique to HIV-1, have been identified in the SIV isolated from the greater spot-nosed monkey (SIVgsn),149 and phylogenetic analyses indicate that SIVgsn (in the env gene) and SIVrcm (in the gag and pol genes) are the two SIVs most closely related to SIVcpz. On the basis of these findings, it has been proposed that SIVcpz arose as a result of a recombination event involving SIVgsn and SIVrcm lineages in a West Central African monkey species upon which chimpanzees prey.28 According to this model, ptt chimpanzees, sharing an overlapping geographical range with greater spot-nosed and red-capped monkeys in West Central Africa, would have been the first member of the great apes to be infected with the putative recombinant progenitor of SIVcpz, eventually giving rise to strains of SIVcpzPtt. The subsequent four cross-species transmissions to humans, thereby generating the M, N, O, and P groups of HIV-1, would be consistent with the tree locations of these primate lentiviruses (Fig. 49.4A).
Genomic Organization of HIVs
Nucleotide sequencing of several of the original HIV-1 isolates revealed an unexpected result: in contrast to the well-characterized and intensively studied prototypical retroviruses such as ASLV and MLV, whose genomes contain only three genes (gag, pol, and env) encoding the structural proteins and enzymes required for productive infection, the HIV-1 genome included several additional and overlapping open reading frames (ORFs) of unknown function (Fig. 49.5). Not only were HIV-1 and HIV-2 shown to contain multiple additional ORFs, but also their genomic organizations appeared to be very similar. Further analyses revealed that HIV-1 contained the distinguishing vpu gene144,726 and HIV-2 carried a signature vpx375 gene.
Like all replication-competent retroviruses, the three primary HIV-1 translation products, all encoding structural proteins or enzymes, are initially synthesized as polyprotein precursors, which are subsequently processed by viral or cellular proteases into mature, particle-associated proteins (Fig. 49.6). The 55-kD Gag precursor Pr55Gag is cleaved into the matrix (MA), capsid (CA), nucleocapsid (NC), p6, and the two spacer peptides SP1 and SP2 during or after the release of progeny virions. Autocatalysis of the 160-kD Gag-Pol polyprotein, Pr160Gag-Pol, gives rise to the protease (PR), the heterodimeric RT, and integrase (IN) proteins, whereas proteolytic digestion by a cellular enzyme converts the glycosylated 160-kD Env precursor, gp160, into the gp120 surface (SU) and gp41 transmembrane (TM)
cleavage products. The remaining six HIV-1–encoded proteins (Vif, Vpr, Tat, Rev, Vpu, and Nef) are the primary translation products of spliced messenger RNAs (mRNAs).
cleavage products. The remaining six HIV-1–encoded proteins (Vif, Vpr, Tat, Rev, Vpu, and Nef) are the primary translation products of spliced messenger RNAs (mRNAs).
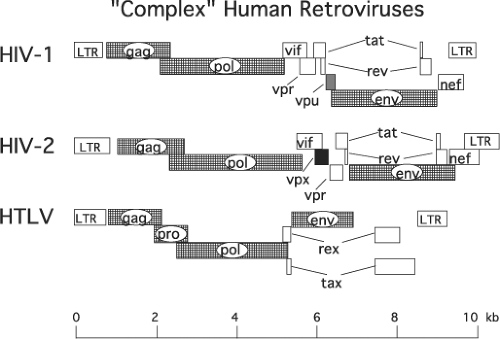 Figure 49.5. Genomic organization of simple and complex retroviruses. The genes of Moloney murine leukemia virus (MLV), human T-cell leukemia virus 1 (HTLV-1), human immunodeficiency virus 1 (HIV-1), and HIV-2 are depicted as they are arranged in their respective proviral DNA. The sizes of the different proviral DNA are shown in proportion to the 9.7-kb HIV provirus. |
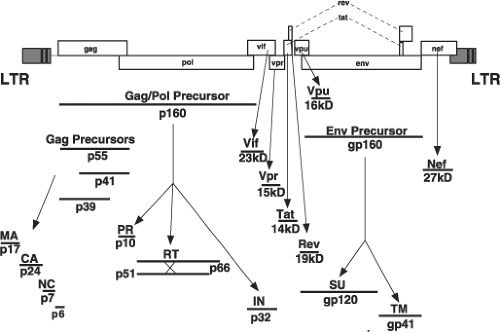 Figure 49.6. Human immunodeficiency virus (HIV)-encoded proteins. The location of the HIV genes, the sizes of primary translation products (in some cases, polyproteins), and the processed mature viral proteins are indicated. |
HIV-1 and HIV-2 have incorporated multiple sequence elements into their genomic RNAs that direct the balanced and coordinated production of progeny virions. Many of these cis-acting RNA elements (Fig. 49.7) are present in other retroviral genomes, but a few are unique to the primate lentiviruses. The 5′ ∼330 nucleotide untranslated region of HIV-1 genomic RNA is highly structured and contains multiple elements that mediate transcriptional elongation of viral RNA transcripts, splicing, genomic RNA dimerization, packaging of full-length viral RNA, and reverse transcription. A schematic model of this 5′ leader sequence, deduced from genetic, functional, structural, and nuclease-accessibility mapping experiments, is shown in Figure 49.7, inset A.
Three regions encompassing the primer binding site (pbs) stem participate in the placement and stabilization of
the transfer (t) RNALys3 primer, which is incorporated into HIV-1 particles and is required for the initiation of reverse transcription. These regions include the pbs itself, which can base-pair with the 3′ terminal 18 nt of tRNALys3; the primer activation signal (PAS), which interacts with the thymidine-pseudouridine-cytidine (TψC) arm of the tRNA to trigger the reverse transcription reaction; and the A-loop, which is complementary to the anticodon loop of tRNALys3 (reviewed in60). The unwinding of both the tRNA primer and the pbs stem within the 5′ untranslated region (UTR) of the HIV-1 genome, as well as the incorporation of the primer into nascent virions, is facilitated by the NC domain of the Gag polyprotein, which plays the role of a molecular chaperone in this process (reviewed in440).
the transfer (t) RNALys3 primer, which is incorporated into HIV-1 particles and is required for the initiation of reverse transcription. These regions include the pbs itself, which can base-pair with the 3′ terminal 18 nt of tRNALys3; the primer activation signal (PAS), which interacts with the thymidine-pseudouridine-cytidine (TψC) arm of the tRNA to trigger the reverse transcription reaction; and the A-loop, which is complementary to the anticodon loop of tRNALys3 (reviewed in60). The unwinding of both the tRNA primer and the pbs stem within the 5′ untranslated region (UTR) of the HIV-1 genome, as well as the incorporation of the primer into nascent virions, is facilitated by the NC domain of the Gag polyprotein, which plays the role of a molecular chaperone in this process (reviewed in440).
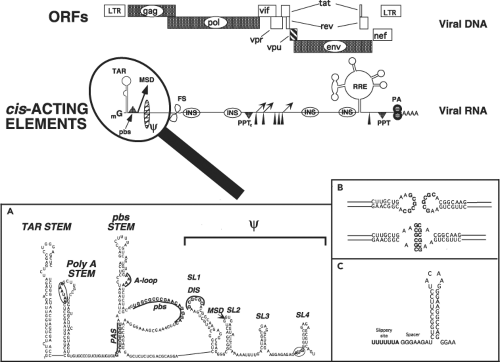 Figure 49.7. Cis–acting elements present in the human immunodeficiency virus 1 (HIV-1) genome. Elements associated with HIV-1 genomic RNA include the methyl-capped terminal G residue at the 5′ terminus of viral RNA (mG), Tat-responsive stem-bulge-loop structure (TAR), binding site for the tRNALys primer (pbs), major splice donor (MSD), RNA packaging site (ψ), frameshifting motif (FS), RNA instability or nuclear retention elements (INSs), central polypurine tract (PPTc), splice acceptors (arrowheads), internal splice donors (small arrows), Rev-responsive element (RRE), canonical 3′ polypurine tract (PPT), and polyadenylation signal (PA). A: The putative secondary structure at the 5′ terminus of HIV-1 messenger RNA (mRNA). The positions of the TAR stem-loop, poly(A) stem-loop, circled poly(A) addition signal (AAUAAA), pbs, dimerization initiation sequence (DIS), MSD, primer activation signal (PAS), ψ stem-loops 1 to 4 (SL1 to SL4), and translation initiation AUG codon for Gag are indicated. (A adapted from Carteau S, Gorelick RJ, Bushman FD. Coupled integration of human immunodeficiency virus type 1 cDNA ends by purified integrase in vitro: stimulation by the viral nucleocapsid protein. J Virol 1999;73:6670–6679.) B: The self-complementary DIS sequences, located at the crown of stem-loop 1, participate in the formation of “kissing loop” intermediates, an initial step in the RNA dimerization reaction. C: RNA stem-loop structure downstream of the UUUUUUA frameshifting sequence. |
The packaging of HIV-1 genomic RNA into progeny virus particles is also directed by the NC domain of the Gag polyprotein and is primarily dependent on its interaction with the four RNA stem-loops (SL1, SL2, SL3, and SL4) constituting the core encapsidation signal, ψ (Fig. 49.7A).157 It is currently thought that a fully functional HIV ψ packaging signal may include the entire 5′ UTR plus the first 300 nucleotides of the gag gene. In virions, the HIV-1 genome is present as an RNA dimer, noncovalently linked at the 5′ end of each molecule. Particle-associated dimeric RNA provides an alternative template for reverse transcription, should the enzyme encounter a nick or break during the polymerization reaction. The dimerization initiation sequence (DIS), located at the crown of SL1, contains
a complementary, exposed, and palindromic sequence (Fig. 49.7, insets A and B).427,704 Most of the non-B/D HIV-1 subtypes, including the highly prevalent subtype C viruses, have a GTGCAC for their DIS, whereas clade B and D subtypes have a GCGCGC in their DIS. Dimerization is initiated via a kissing loop mechanism involving base-pairing between the palindromic sequences present on paired HIV-1 genomes.579 Other regions of SL1 participate in stabilizing the RNA dimer into a more extended duplex structure,207 a process also believed to be chaperoned by NC. The ψ packaging domain also contains the major splice donor (MSD) (near the crown of SL2) and the AUG Gag start codon (at the base of SL4).
a complementary, exposed, and palindromic sequence (Fig. 49.7, insets A and B).427,704 Most of the non-B/D HIV-1 subtypes, including the highly prevalent subtype C viruses, have a GTGCAC for their DIS, whereas clade B and D subtypes have a GCGCGC in their DIS. Dimerization is initiated via a kissing loop mechanism involving base-pairing between the palindromic sequences present on paired HIV-1 genomes.579 Other regions of SL1 participate in stabilizing the RNA dimer into a more extended duplex structure,207 a process also believed to be chaperoned by NC. The ψ packaging domain also contains the major splice donor (MSD) (near the crown of SL2) and the AUG Gag start codon (at the base of SL4).
It has been proposed that the HIV-1 leader can switch from the multiple hairpin structure shown in Figure 49.7A to an alternate, rod-like conformation, and that each conformer specifically promotes either packaging or translation of the viral RNA.340 In this model, the individual stem-loops are formed as intermediates during RNA synthesis, but the entire HIV-1 leader is eventually converted to the thermodynamically more stable rod structure containing an exposed AUG Gag start codon and an occluded DIS motif. The rod conformer, containing a masked DIS, may promote the translation of HIV-1 mRNAs into viral-encoded proteins. A subsequent switch to the alternative conformation, mediated by NC, would expose the DIS and ψ elements, favoring dimerization, packaging, and, ultimately, reverse transcription of the viral genomic RNA.160,575 Evidence for the existence of alternative RNA conformers in virions or infected cells and the hypothesized conformational switch is still forthcoming.
In HIV-1, the AAUAAA binding site for the cleavage and polyadenylation factor (CPSF) and the GU/U-rich binding site for the 3′ terminal cleavage stimulation factor (CstF) are both situated within the repeat (R) regions at each end of the viral genomic RNA. An active 5′ poly(A) site would obviously compromise the generation of long RNA transcripts by mediating premature cleavage and polyadenylation. A polyadenylation enhancer, situated upstream of the 3′ poly(A) signals, stabilizes the binding of CPSF to AAUAAA and promotes poly(A) addition, whereas the 5′ poly(A) stem or the closely positioned MSD has been reported to suppress polyadenylation activity.25,161,258
Lentiviruses are unique among retroviruses in having a novel central polypurine tract (PPT) in addition to the canonical U3-proximal PPT, both of which are used for the synthesis of plus-strand viral DNA.114 The viral genomic RNA also contains a heptameric UUUUUUA “slippery” sequence within the gag gene where ribosomal frameshifting (FS) occurs. This sequence functions in conjunction with a downstream 8-nt spacer and hairpin (Fig. 49.7C) to mediate minus 1 translational frameshifting.353 The viral RNA also folds into two additional complex structures (the transactivation response region [TAR] and Rev-responsive element [RRE]), which are involved in RNA synthesis and RNA nuclear-to-cytoplasmic transport, respectively. Multiple inhibitory sequences (INSs), associated with the instability or nuclear retention of HIV transcripts, are scattered throughout genes encoding the gag, pol, and env genes.480,640 Finally, although not strictly considered a cis-acting element, HIV-1 genomic RNA contains a significantly higher adenosine deoxyribonucleotide (A) content (approximately 39%) than does mammalian DNA; this bias contributes to an unusual codon usage.51
The secondary structure of the entire HIV-1 genome has recently been determined using a technique called selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE).791 SHAPE is based on the chemical modification of unpaired nucleotides in RNA molecules and the resultant termination of reverse transcription at the altered site. Regions of RNA with significant secondary structure are not readily modified, whereas looped-out RNA segments exhibit high SHAPE reactivity. This allows the determination of the folded state of RNA using a computer algorithm.503 Some of the tightly base-paired RNA regions identified by SHAPE correspond to previously described TAR structures at the 5′ and 3′ termini of the HIV-1 genome, the SP1 encapsidation site in the 5′ UTR, and the RRE (Fig. 49.7). Interestingly, structured RNA segments were also present in protein-coding regions located between individual Gag, Pol, and Env subdomains, which give rise to mature protein cleavage products. In addition, base-paired regions of HIV-1 RNA are located adjacent to sequences encoding protein domains that undergo co-translational folding or unfolding.791 Ribosomal pausing induced at these structured RNA segments may retard translation to allow sufficient time for a nascent protein to attain its final conformation. Nonstructured RNA regions encoding the gp120 variable loops were enclosed by stable RNA duplexes, yet another example of co-related RNA and protein structures.
biology of HIV infections
The main cellular targets for HIV-1 in infected individuals are the CD4+ T lymphocytes, CD4+ cells of the macrophage lineage, and some populations of dendritic cells (DCs). A quintessential property of HIV-1 and the other primate lentiviruses is to sequentially use CD4 and a second cellular receptor during entry into susceptible cells, a subject discussed more extensively in the section dealing with virus binding and entry. The earliest studies used mitogen-activated human PBMCs for HIV-1 isolation and propagation. Some of the original HIV-1 isolates were also able to infect continuous CD4+ T leukemia cell lines such as CEM, Jurkat, Hut-78 (including its H9 derivative), and the HTLV-1–containing MT-2 and MT-4 cell lines. Human T-cell lines were obviously more logistically tractable than PBMCs and their use greatly facilitated early molecular and genetic studies of HIV-1.
Replication and Tropism
HIV-1 isolates were initially classified on the basis of their replicative, cytopathic, and tropic properties. A majority of primary virus isolates, which replicated slowly and generated small amounts of progeny particles, were labeled “slow/low” to distinguish them from “rapid/high” HIV-1 strains, which exhibit faster infection kinetics and release high titers of virions. Some HIV-1 primary isolates, particularly those recovered from symptomatic individuals, induced syncytium formation (see Fig. 49.2B). These isolates were designated syncytium-inducing (SI) to distinguish them from non–syncytium-inducing (NSI) strains. Some SI isolates are able to infect CD4+ T-cell lines but not monocyte-derived macrophages (MDMs) and have been designated T-cell-line or TCL-tropic viruses. Cell tropism was a third property used to classify HIV-1 primary isolates. All HIV-1 strains are able to productively infect activated PBMCs;
some can also replicate in cultures of MDMs and have been classified as macrophage- or M-tropic. Other primary HIV-1 isolates are also able to infect both MDMs and T-cell lines; such strains have been classified as dual-tropic. Although exceptions exist, M-tropic isolates are usually NSI and “slow/low,” whereas TCL-tropic strains are frequently SI and exhibit the “rapid/high” replication phenotype. Forced passage of primary isolates in continuous CD4+ human T-cell lines has given rise to T-cell-line–adapted (TCLA) HIV-1 strains, which, in contrast to their progenitors, possess increased sensitivity to both neutralizing antibodies and soluble CD4.730
some can also replicate in cultures of MDMs and have been classified as macrophage- or M-tropic. Other primary HIV-1 isolates are also able to infect both MDMs and T-cell lines; such strains have been classified as dual-tropic. Although exceptions exist, M-tropic isolates are usually NSI and “slow/low,” whereas TCL-tropic strains are frequently SI and exhibit the “rapid/high” replication phenotype. Forced passage of primary isolates in continuous CD4+ human T-cell lines has given rise to T-cell-line–adapted (TCLA) HIV-1 strains, which, in contrast to their progenitors, possess increased sensitivity to both neutralizing antibodies and soluble CD4.730
Table 49.1 Tropic and Biological Properties of HIV-1 Isolates | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
The discovery that α and β chemokine receptors function as co-receptors (in addition to CD4) for virus entry127,181,216 led to a revised classification system for HIV-1 strains (Table 49.1). Isolates are now categorized on the basis of chemokine receptor usage in fusion assays, in which CD4 and an individual co-receptor protein are expressed in reporter cells, or in entry assays, in which small-molecule co-receptor–targeted inhibitors, specific for CCR5 or CXCR4, block single-cycle or spreading viral infections in T-cell lines or human PBMCs. As indicated in Table 49.1, HIV-1 isolates using the CXCR4 receptor (now designated X4 viruses47) are frequently TCL-tropic, SI strains that exhibit enhanced cytopathicity and more vigorous replicative properties. In contrast, HIV-1 strains exclusively using the CCR5 receptor (R5 viruses) may be M-tropic and NSI. Dual-tropic R5/X4 strains exhibiting a continuum of tropic phenotypes are frequently SI. Interestingly, most T-cell leukemia cell lines express high levels of CXCR4 but are CCR5 negative, explaining in part why primary HIV-1 isolates fail to replicate in these cells.
HIV-1 Infection of CD4+ T Lymphocytes
CCR5-using viruses are the predominant HIV-1 strains detected in recently infected individuals and remain the dominant virus throughout the asymptomatic phase of HIV-1 infection. In vivo, CCR5 is exclusively expressed on memory CD4+ T cells, a subset present at high levels in nonlymphoid tissues (e.g., in the gastrointestinal tract and lung).191 Many of these tissue-infiltrating memory CD4+ T cells are also activated. In contrast, CXCR4 is expressed on both naïve and memory CD4+ T-cell populations. In peripheral blood, where a majority of the circulating lymphocytes are naïve, 80% to 95% of the CD4+ T cells may express CXCR4, whereas only 5% to 10% produce detectable CCR5.280,560 Because R5 HIV-1 strains predominate in recently infected individuals, marked depletion of CCR5-expressing memory CD4+ T lymphocytes in the gastrointestinal tract has been reported, whereas more modest reductions of total CD4+ T-cell numbers in the blood occur during the acute infection.516 The emergence of X4 variants during the symptomatic phase of infection, which occurs in approximately 50% of individuals infected with clade B HIV-1, is referred to as co-receptor switching. It is associated with accelerated CD4+ T-cell depletion and more rapid progression to AIDS. Such infected individuals have 30- to 70-fold higher frequencies of virus-infected naïve cells than persons who harbor only R5 virus, consistent with the co-receptor usage of each HIV-1 strain.64 The accelerated and specific decline of the naïve CD4+ T cells after the emergence of X4 strains represents an additional insult to the immune system that severely impedes CD4+ T-lymphocyte renewal and heralds the beginning of a downhill clinical course.
Studies of acute HIV and SIV/SHIV infections have shown that nonactivated CD4+ T lymphocytes in the mucosal lamina propria and draining lymph nodes are the principal virus targets during this phase of the infection.447,562,851 Paradoxically, the innate immune responses directed against HIV following virus acquisition induce a cascade of inflammatory cytokines that greatly increases the numbers and activation status of CD4+ T cells. In contrast to these findings, multiple studies have shown that resting PBMCs in culture are highly refractory to HIV-1 infection; no viral proteins or progeny virions are produced.718,843 PBMCs, activated with mitogens or following incubation with anti-CD3 and anti-CD28 monoclonal antibodies (mAbs) and propagated in the presence of interleukin-2 (IL-2), are quite susceptible to HIV-1 and produce high titers of infectious virus. The block to HIV-1 replication in noncycling cultured PBMCs is not presently understood. Indeed, cell cycle studies have shown that progression into G1b is required to achieve productive HIV-1 infections.405 In vivo, activated CD4+ T lymphocytes are the main source of progeny virions. Nonetheless, an inducible reservoir of resting “latently infected” memory CD4+ T cells, containing integrated HIV-1 DNA and expressing no viral proteins, is established in exposed individuals within days of transmission and is thought to be the driving factor preventing the eradication of this viral infection.135,137
During infections of human T-cell lines or CD4+ T lymphocytes, HIV-1 can spread through the cultures by two different mechanisms: (a) fluid phase diffusion of cell-free particles or (b) virological synapse (VS)-mediated cell-to-cell contact. Early studies reported that cell-to-cell transmission of HIV-1 was 102– to 103-fold more efficient than that of cell-free virus.184 The extremely low particle-to-infectivity ratio (10−3 to 10−5) for HIV-1 undoubtedly contributed to the inefficient spread of cell-free virions. The formation of a VS with T cells, first reported for HTLV-1,344 utilizes some of the components of the immunologic synapse machinery involved in the interaction between lymphocytes and antigen-presenting cells.709 The formation of the HIV-1 VS is initiated by the binding of
gp120 in the infected cell to CD4 molecules on the surface of the target cell.365 This conjugate is further stabilized by interactions between leukocyte function–associated antigen (LFA) and intercellular adhesion molecules (ICAMs), which promote additional recruitment of actin, viral proteins (Gag and Env), CD4, and integrins to cell–cell contact regions. Cytoskeletal remodeling drives virus assembly in the infected cell to the plasma membrane and toward the target cell. Progeny particles released into the synaptic space enter the recipient cell by fusion with the plasma membrane or following passage through endosomal compartments.660 The co-polarization of clustered viral proteins in the donor cell with receptors in multiple adjacent recipients can also lead to the formation of polysynaptic structures.366 Filopodial bridges and other actin-containing tubular extensions of the plasma membrane have been reported for delivering virions to distant target cells.693 Inhibitors of HIV-1, including neutralizing antibodies, are able to block cell-free and cell-to-cell–mediated virus infectivity, although transfer across the VS may offer some protection from these agents.121
gp120 in the infected cell to CD4 molecules on the surface of the target cell.365 This conjugate is further stabilized by interactions between leukocyte function–associated antigen (LFA) and intercellular adhesion molecules (ICAMs), which promote additional recruitment of actin, viral proteins (Gag and Env), CD4, and integrins to cell–cell contact regions. Cytoskeletal remodeling drives virus assembly in the infected cell to the plasma membrane and toward the target cell. Progeny particles released into the synaptic space enter the recipient cell by fusion with the plasma membrane or following passage through endosomal compartments.660 The co-polarization of clustered viral proteins in the donor cell with receptors in multiple adjacent recipients can also lead to the formation of polysynaptic structures.366 Filopodial bridges and other actin-containing tubular extensions of the plasma membrane have been reported for delivering virions to distant target cells.693 Inhibitors of HIV-1, including neutralizing antibodies, are able to block cell-free and cell-to-cell–mediated virus infectivity, although transfer across the VS may offer some protection from these agents.121
Although HIV-1 causes the loss of CD4+ cells in culture and depletion of this T-lymphocyte subset in virus-infected individuals, the mechanisms responsible for this cytopathic effect are still incompletely understood. HIV-1–induced killing of cultured CD4+ T cells (primary T cells and T-cell lines) occurs by necrosis or cytolysis rather than by classical apoptosis, which involves the activation of cysteinyl aspartate-requiring proteases (known as caspases).72 Some viral-encoded proteins including Vpr and Env are cytotoxic when expressed alone or in the context of a productive HIV infection. In vivo, the massive depletion of CD4+ T lymphocytes in blood and tissues during acute HIV and SIV infections is now recognized to be the result of direct virus-induced cell killing.506,516 Nonetheless, apoptotic CD4+ T lymphocytes are present in the peripheral blood and lymph nodes of HIV-1 seropositive individuals during the chronic phase of infection. These cells are uninfected and are thought to represent the effects of bystander cell damage due to the unrelenting and generalized immune activation, a major factor contributing to HIV-1 pathogenesis.282
HIV-1 Infection of Macrophages and Dendritic Cells
All known lentiviruses have retained the capacity to replicate in nonproliferating, terminally differentiated tissue macrophage. Unlike the oncoretroviruses, which require mitosis and dissolution of the nuclear membrane to complete a cycle of replication, HIV-1 is able to productively infect G1 (but not G0)-arrested macrophages. This property requires that the HIV-1 preintegration complex (PIC) be translocated through intact nuclear pores, a process in which the capsid (CA) protein modulates interactions with the nuclear transport machinery in nondividing cells432 (see section on Nuclear Import). In HIV-1–positive individuals, infected macrophages contribute to viral persistence and dissemination to the central nervous system (CNS). HIV-1 is thought to enter the CNS very soon following transmission as virus-free particles or by the trafficking of infected lymphocytes (and possibly infected monocytes). HIV-1 does not productively infect neurons but instead targets perivascular macrophages in the choroid plexus and parenchymal microglial cells. Multinucleated giant cells, suggestive of virus-induced fusion of microglia and/or macrophages, are a histopathologic characteristic of HIV-1–associated neuropathy. Neurologic dysfunction affecting the CNS and the peripheral nervous system is observed in 40% to 70% of virus-positive individuals. HIV-1 replication in monocyte/macrophage lineage microglia within the CNS may persist in patients receiving HAART because of inefficient transfer of drugs across the blood–brain barrier. This viral reservoir, as well as HIV-1 present in latently infected memory CD4+ T cells, could contribute to the rebound of plasma viremia invariably observed after cessation of antiretroviral drug administration.
Virtually all information pertaining to HIV-1 replication in macrophages derives from studies of an in vitro surrogate: monocyte-derived macrophage (MDM). The vigorous replication by M-tropic viral strains in MDM requires that blood-derived monocytes undergo differentiation in vitro.682 MDMs and tissue macrophages express lower surface levels of CD4, CCR5, and CXCR4 than CD4+ T lymphocytes.441,788 Nonetheless, only M-tropic R5 HIV-1 strains are able to initiate infections of MDM and only a subset of M-tropic viruses are also neurotropic.326 Most primary HIV-1 isolates from blood are weakly M-tropic; strong macrophage tropism is mainly seen with variants from tissues such as the CNS, where macrophage dependence is paramount.598 Like productive infections of T lymphocytes, HIV-1 replication in MDMs requires integration of the reverse transcript.206 However, in contrast to numerous reports demonstrating that assembly and release of progeny virions occur at the plasma membrane of productively infected T cells, electron microscopy of HIV-1–infected MDM revealed accumulations of particles within intracellular vacuoles of endosomal origin.576,589 This interpretation has been questioned by reports showing that particles within putative internal vesicles were in a compartment, distinct from multivesicular bodies, which communicates with the exterior through tubular channels continuous with the highly invaginated macrophage plasma membrane.45,180,798 In fact, a recent study employing electron tomography and electron microscopy with stereology has reported that MDMs release progeny virions into a pre-existing membranous compartment, contiguous with the plasma membrane, which expands in size during HIV-1 infection.797 Virus particles that assemble in this internal compartment in MDMs are rapidly translocated to the VS following contact with susceptible T cells.277
DCs are antigen-presenting cells that capture, transport, and present antigens to CD4+ and CD8+ T lymphocytes. Two main subtypes of DCs have been identified: myeloid DCs (MDCs) and plasmacytoid DCs (PDCs). MDCs reside in multiple tissues including the skin (where they are called Langerhans cells) and the intestinal and genital tract mucosa. The less abundant PDCs are found in the blood, T-cell zones of lymph nodes, and thymus and can be recruited to sites of inflammation. After activation, MDCs secrete IL-12, whereas PDCs produce type I interferons and inflammatory cytokines. MDCs present near mucosal surfaces may represent the first line of defense against sexually transmitted HIV-1, transporting virus particles from this portal of entry to draining lymph nodes where they are degraded into antigenic peptides for presentation to CD4+ T lymphocytes. Paradoxically, this critical initial response exacerbates the HIV-1 acute infection by delivering virus to the very T-cell subset that is susceptible to and can massively amplify progeny virus production. Because of the low abundance of DCs in tissues and the small number of these cells recoverable from clinical specimens, most studies evaluating the interaction
of DCs with HIV-1 have used monocytes differentiated in the presence of granulocyte-macrophage colony-stimulating factor (GM-CSF) and IL-4. These derivatives are known as monocyte-derived DCs (MDDCs).
of DCs with HIV-1 have used monocytes differentiated in the presence of granulocyte-macrophage colony-stimulating factor (GM-CSF) and IL-4. These derivatives are known as monocyte-derived DCs (MDDCs).
Productive HIV-1 infections of MDDCs are very inefficient compared to infections of activated CD4+ T cells, although vigorous virus replication is observed when DCs are first pulsed with virus and then co-cultivated with CD4+ T lymphocytes, a process called trans-infection.259 Immature DCs (IDCs) express relatively low levels of CD4, CCR5, and CXCR4 and larger amounts of C-type lectin receptors (CLRs) such as dendritic Cell-Specific Intercellular adhesion molecule-3-Grabbing Non-integrin (DC-SIGN) and dendritic cell immunoreceptor (DCIR), which capture HIV by binding to gp120-associated high-mannose glycans. Functionally, maturation of DCs leads to (a) increased capacity to degrade and present antigen to T cells by forming immunologic synapses, (b) augmented secretion of T-cell co-stimulatory molecules, and (c) potent suppression of endolysosomal degradation. Cell surface CLR expression, including that of DC-SIGN, is markedly reduced in mature DCs; capture of HIV-1 is now considered to be CLR, gp120, and CD4 independent in mature DCs.
Early studies reported that HIV-1 was internalized in trypsin-resistant, low-pH compartments for several days in MDDCs or cells expressing DC-SIGN.415 Co-cultivation with activated CD4+ T cells resulted in rapid trans-infection.259 EM analyses of infected MDDCs indicated that intact HIV particles were present within cytoplasmic vesicles. Subsequent imaging studies, using green fluorescent protein (GFP)-labeled HIV, revealed that DCs transferred virus to T cells via a VS, utilizing adhesion molecules similar to those used in the formation of the immunologic synapse.415 Virions in the infected DC were co-polarized with CD4 and chemokine co-receptors in the T lymphocyte at the cell interfaces. More recent experiments have shown that in DCs, HIV-1 resides in a unique surface-accessible intracellular compartment, distinct from endocytic vesicles, and can rapidly traffic to the VS following contact with T cells.837 This unusual DC compartment communicates with the extracellular environment and, similar to structures present in HIV-1–infected macrophages, is composed of highly folded invaginations of the plasma membrane.45,798 DC-SIGN/gp120-mediated HIV-1 entry into IDCs by endocytosis very likely results in extensive degradation of viral proteins by an active endocytic system primed for antigen presentation. Consequently, IDCs would only transmit residual nondegraded HIV remaining on their surfaces to T lymphocytes. In contrast, mature DCs, unable to endocytose HIV in a normal fashion, sequester virions in specialized intracellular compartments and, during antigen presentation, rapidly transfer infectious particles to CD4+ T cells. Recent studies indicate that HIV-1 infection of DCs, and to a lesser extent mature DCs, is restricted by the interferon-induced host factor SAM domain and HD domain–containing protein 1 (SAMHD1).46,328,418 The ability of SAMHD1 to interfere with lentiviral infection is counteracted by Vpx (see section on Vpx).
HIV-1 Animal Models
Chimpanzees
In the search for an HIV-1 animal model during the early phase of the AIDS epidemic, cell suspensions from virus-infected individuals were inoculated into a variety of mammalian species including nonhuman primates, but only chimpanzees consistently became infected. It was subsequently shown that asymptomatic HIV-1 infections could readily be established in chimpanzees, but viremia was not maintained and no long-standing impairment to the immune system occurred.24 This could reflect genetic changes, which occurred in the SIVcpz progenitor of HIV-1 following its transmission from chimpanzees and adaptation to humans.663 The failure of chimpanzees to develop disease in a timely fashion following inoculation with tissue culture–adapted or patient-derived HIV-1 isolates, coupled with their endangered species status, has curtailed their use as an animal model of HIV-1–induced immunodeficiency.
Transgenic Mice
When it became apparent that HIV-1 infections could only be established in chimpanzees and humans, potentially more tractable and novel rodent systems were developed to model specific steps of in vivo HIV-1 infections. In one approach, transgenic mice containing the entire HIV-1 genome or individual viral genes under the control of the HIV-1 long terminal repeat (LTR) were constructed. Because mice carrying such transgenes contain HIV-1 DNA in every cell, they model the postintegration phase of the virus life cycle and provide a means for assessing possible deleterious effects of viral proteins in vivo. In general, these first-generation transgenic animals expressed very low levels of HIV-1 proteins and occasionally developed disease.
To overcome the intrinsically low levels of HIV-1 LTR-directed expression in rodents, transgenic mice were subsequently generated carrying viral genes under the control of cellular/tissue-specific promoters. In particular, mice carrying a complete copy of the HIV-1 genome or just the nef gene, directed by a chimeric (human-mouse) CD4 promoter/enhancer element, spontaneously developed an AIDS-like disease characterized by marked depletion of CD4+ T lymphocytes, thymic atrophy, T-cell activation, and immunodeficiency.292,618 Thus, targeting HIV-1 Nef synthesis to mouse CD4+ cells, in the absence of a spreading viral infection or the expression of other HIV-1 gene products, was sufficient to elicit a disease with some of the features of human AIDS.
Xenotransplantation Mouse Models
In addition to their use for investigations of human hematopoiesis, innate and adaptive immunity, cancer biology, and regenerative medicine, mice engrafted with human cells have been utilized as a small-animal model for studies of HIV-1 pathogenesis and vaccine development. In very early studies, severe combined immunodeficiency (SCID) mice, engrafted with human PBMC or fragments of human fetal thymus and fetal liver engrafted under the renal capsule, were shown to support HIV-1 replication and moderate to profound depletions of CD4+ T lymphocytes but generated no immune responses to the virus.543,554 Relatively low levels of human cell engraftment were achieved in these first-generation humanized mice.
Although the subsequent development of nonobese diabetic (NOD) SCID animals resulted in improved human PBMC engraftment, the relatively short life span of the mice and residual murine natural killer (NK) cell activity limited their usefulness. Targeted mutations of the mouse interleukin-2 receptor-γ chain (γc−/−) and reconstitutions with PBMC, hematopoietic stem cells (HSCs), or human cord blood greatly increased engraftment of human tissue.348 Inoculation of these mice with HIV-1 by parenteral and mucosal routes
resulted in sustained disseminated infections.27,49 The recently described surgical implantation of fetal thymic and liver tissues in humanized mice previously engrafted with HSCs resulted in animals able to support high levels of HIV viremia, experience CD4+ T-cell depletion, sustain generalized immune activation, and generate virus-specific antibody and CD4+/CD8+ T-cell responses.81 Although great progress has been made in the development of xenotransplant mouse models over the past 20 years, they are technically demanding to make and not currently amenable for widespread use.
resulted in sustained disseminated infections.27,49 The recently described surgical implantation of fetal thymic and liver tissues in humanized mice previously engrafted with HSCs resulted in animals able to support high levels of HIV viremia, experience CD4+ T-cell depletion, sustain generalized immune activation, and generate virus-specific antibody and CD4+/CD8+ T-cell responses.81 Although great progress has been made in the development of xenotransplant mouse models over the past 20 years, they are technically demanding to make and not currently amenable for widespread use.
SIV/HIV Chimeric Viruses
When it became apparent that humans and chimpanzees were the only mammalian species that could be infected by HIV-1, attention turned to the SIV/Asian macaque model with the idea of constructing a virus containing both SIV and HIV gene segments. The earliest versions of these SIV/HIV chimeric viruses (SHIVs) consisted of the genetic backbone of SIVmac239, into which the HIV-1 tat, rev, vpu, and env and, in some instances, portions of vpr and nef genes were inserted (e-Fig. 49.1). SHIVs were constructed because, unlike SIVs, they could be used as challenge viruses in vaccine experiments to monitor the effectiveness of immunogens eliciting antiviral neutralizing antibodies directed against the HIV-1 Env glycoprotein. The first pathogenic SHIVs developed all carried X4-tropic env genes and caused a rapid, systemic, and complete loss of CD4+ T lymphocytes within a few weeks of inoculation, high and sustained levels of viremia (>107 RNA copies/mL of plasma), and death from immunodeficiency within 3 to 6 months of inoculation.345,361,622 This aggressive disease phenotype appeared to be ideally suited for vaccine studies and, for a few years (2000 to 2004), SHIVs rather than SIVs became the challenge virus of choice for such experiments. However, concern was raised when the results from many studies showed that although most vaccine regimens suppressed SHIV replication and disease induction, they failed to protect monkeys from acquisition of either SHIV or SIV infection. In addition, differences were demonstrated between the chemokine receptor utilization of SIV (CCR5) and most transmitted HIV-1 (CCR5) versus the early SHIVs (CXCR4) and the CD4+ T-cell subsets targeted by these viruses in vivo (memory [SIV and HIV-1] versus naïve [SHIV]).563 The development and use of R5-tropic SHIVs as challenge viruses in macaques were shown to be better surrogates for HIV-1 infections of humans.298
Derivatized HIVs (also known as minimal SHIVs), containing more than 90% of the HIV-1 genome and the SIV vif gene (to counteract macaque APOBEC3 proteins) plus a seven–amino acid segment of the SIV Gag analog of the HIV-1 cyclophilin A (CypA)-binding loop (to escape macaque TRIM5α [for tripartite motif-containing α] restriction), have recently been reported to establish transient infections in pig-tailed macaques.346 When it was subsequently appreciated that pig-tailed macaques lacked TRIM5α restriction activity against HIV-1, derivatized HIVs, containing only the SIV vif gene, were made and shown to transiently infect this monkey species.303
Molecular Biology of HIV-1 Replication
Overview
The HIV-1 replication cycle (Fig. 49.8) begins with the binding of virus particles to CD4 molecules on the surface of susceptible cells. In the context of tissue culture systems, susceptible cells include activated CD4+ T lymphocytes present in PBMC collected from uninfected individuals, T-cell leukemia cell lines, and MDMs. In vivo, activation of CD4+ T cells is not required for HIV-1 replication. Although binding of virions to CD4 is essential for HIV infectivity, their subsequent interaction with a co-receptor, now known to be members of the seven-membrane-spanning CC or CXC families of chemokine receptors, is required for membrane fusion and entry (reviewed in13). Multiple chemokine receptors exhibit activity in fusion/entry assays with HIV-1 gp120 (see later section on Env); however, the two most physiologically important co-receptors are CXCR4 and CCR5. Unlike some other enveloped viruses that enter cells by receptor-mediated endocytosis, HIV-1 and many other retroviruses fuse directly with the plasma membrane. Once inside the cell, HIV-1 encounters a cortical actin barrier, which can be remodeled by the virally encoded Nef protein. Partially uncoated virions migrate from cortical actin to the microtubule network where reverse transcription is initiated and movement to the nucleus begins (Fig. 49.8, steps 2 to 4) (reviewed in18). As with other retroelements, the partially double-stranded DNA reverse transcription product is transported through the cytoplasm and to the nucleus as a component of a PIC, which contains a subset of the Gag and Pol proteins. As noted earlier, the lentiviruses are unique among retroviruses in generating PICs that are actively transported by the nuclear import machinery into the interphase nucleus of nondividing cells arrested in the G1 phase of the cell cycle (Fig. 49.8, step 5). After the import of the PIC into the nucleus, full-length linear copies of the reverse transcript are integrated into the chromosomal DNA of the infected cell (Fig. 49.8, step 6), a step required for efficient viral RNA synthesis and infectious particle production.
In activated T lymphocytes, integrated copies of HIV DNA serve as templates for RNA polymerase II (Pol II)-directed viral RNA synthesis (Fig. 49.8, step 7). The coordinated interaction of the HIV-encoded Tat protein and the cellular NF-κB and Sp1 transcriptional transactivating proteins with the RNA Pol II transcriptional apparatus ensures the production of high levels of viral RNA. Unspliced or partially spliced HIV transcripts are exported from the nucleus to the cytoplasm by a unique transport mechanism mediated by the virus-encoded Rev protein, which shuttles between these compartments (Fig. 49.8, step 8). The subsequent translation of the gp160 Env precursor occurs in the endoplasmic reticulum, whereas the Gag and Gag-Pol polyproteins are synthesized on free cytoplasmic ribosomes, and each is independently transported to the plasma membrane (Fig. 49.8, steps 9 and 10). The Gag and Gag-Pol polyproteins, in association with dimers of genomic RNA, condense at the plasma membrane to form an electron-dense “bud” (see Fig. 49.2) that gives rise to a spherical immature particle containing the mature TM and SU Env glycoproteins. Proteolytic processing of the Gag and Pol proteins by HIV PR during or immediately after particle release generates the cone-shaped core characteristic of mature HIV virions (Fig. 49.8, steps 11 and 12).
Thus, HIV-1 uses the same replicative strategy as the so-called simple retroviruses, which encode only the Gag, Pol, and Env proteins. However, during its evolution, HIV-1 has acquired six additional genes to carry out functions that are either (a) performed by cellular proteins already present in the cells infected by the simple retroviruses or (b) uniquely required for virus replication, transmission, and survival in hematopoietic
cells targeted by the primate lentiviruses. Some of the HIV-1 accessory proteins (Vif, Vpr, Vpu, and Nef) are not required for replication in certain human T-cell lines, although virus infectivity may be affected up to several thousandfold, depending on the accessory gene and the type of infected cell. In view of their conservation, however, these additional genes must be required for successful HIV-1 infectivity in vivo.
cells targeted by the primate lentiviruses. Some of the HIV-1 accessory proteins (Vif, Vpr, Vpu, and Nef) are not required for replication in certain human T-cell lines, although virus infectivity may be affected up to several thousandfold, depending on the accessory gene and the type of infected cell. In view of their conservation, however, these additional genes must be required for successful HIV-1 infectivity in vivo.
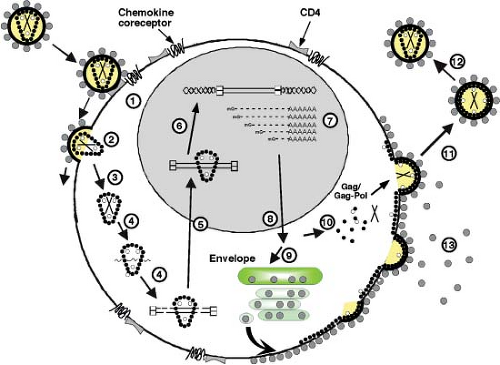 Figure 49.8. The human immunodeficiency virus type 1 (HIV-1) replication cycle. Productive HIV-1 infections begin with the adsorption of cell-free virions to cells and their interactions with both the CD4 and chemokine co-receptors (step 1). In the case of HIV, virus entry (step 2) is a pH-independent process that occurs after the fusion of viral and cellular membranes and results in the partial uncoating (step 3) of incoming virions. Reverse transcription occurs within subviral particles in the cytoplasm of infected cells (step 4), and the double-stranded DNA product within this preintegration complex (PIC) is transported into the nucleus (step 5), where integration into chromosomal DNA (step 6) is mediated by the virus-encoded integrase (IN) (open circles). The integrated viral DNA serves as a template for DNA-dependent RNA polymerase (Pol II) and leads to the production of messenger RNAs (mRNAs) (step 7), some of which are spliced and exported into the cytoplasm (step 8), where they are translated into viral proteins. Envelope (step 9) and Gag plus Gag-Pol (step 10) polyproteins are transported via independent pathways to the plasma membrane, where progeny virus particles begin “budding” from cells and are released as immature particles (step 11). Subsequent proteolysis by the virion-encoded protease (PR) generates mature particles (step 12) containing a characteristic condensed core. Non–virion-associated gp120 Env glycoprotein is also released from cells (step 13). |
HIV-1 encodes only 15 proteins and like all other viruses must utilize a large number of cellular proteins for successful replication. Over the years, several techniques, such as immunoprecipitation/mass spectrometry and yeast two-hybrid screening, have been used to identify such host cell factors. The recent development of genome-wide small-interfering RNA (siRNA) or short hairpin (shRNA) screening has provided genetic approaches to identify potential human proteins required for HIV-1 replication, based on loss of function readouts.82,404,836,857 In these experiments, siRNAs are transfected into target cells, whereas shRNAs are delivered using lentiviral vectors and subsequently processed into siRNAs intracellularly.
Of the 842 genes identified as being required for HIV-1 replication in three siRNA screens, 34 were also reported in at least two screens and three were detected in all three screens. The overlap between the three siRNAs and the single shRNA screen ranged from zero to three genes in pairwise comparisons. Eleven of the 34 genes found in two siRNA screens had also been previously reported to encode HIV-1–interacting proteins and 29 of the 34 genes are expressed in cells susceptible to HIV-1 (CD4 and chemokine coreceptor positive). The overlap between individual screens is unexpectedly small yet is statistically significant.92 This result is not surprising given that different challenge viruses, cell lines, siRNA libraries, periods of gene silencing prior to infection, and readouts of infection were used in the four studies. Nonetheless, all three siRNA screens identified factors associated with transport through the nuclear pores, trafficking between cellular compartments, DNA repair, RNA binding, and interaction with ubiquitin/proteasomal degradation machinery.92 Two of the siRNA screens identified previously reported cellular proteins known to be critical for HIV-1 infectivity including CD4, CXCR4, RelA, elongin B, and cyclin T1. Cyclophilin A, Sp1, and lens epithelium-derived growth factor (LEDGF) are among the known HIV-1 co-factors that were not indentified in any of the screens.
Virus Binding and Entry: The Env Glycoproteins
The HIV Env glycoproteins are synthesized from the singly spliced 4.3-kb Vpu/Env bicistronic mRNA (see Fig. 49.6); translation occurs on ribosomes associated with the rough endoplasmic reticulum (RER). The Env glycopolyprotein precursor, gp160, is an integral membrane protein that is anchored to cell membranes by a hydrophobic stop-transfer signal in the gp41 TM domain (Fig. 49.9) (reviewed in117). gp160 is co-translationally glycosylated and undergoes oligomerization in the endoplasmic reticulum (ER). The predominant, biologically active oligomeric form is a trimer. gp160 is transported to the Golgi, where, like other retroviral Env precursor glycoproteins, it is proteolytically cleaved by a cellular enzyme to the mature SU and TM glycoproteins, gp120 and gp41, respectively (Fig. 49.9). The cellular enzyme primarily responsible for cleavage of retroviral Env precursors following a highly conserved Lys/Arg-X-Lys/Arg-Arg motif is furin or a furin-like protease.288 Cleavage of gp160 is strictly required for Env-induced fusion activity and virus infectivity.237,510 Subsequent to gp160 cleavage, gp120 and gp41 form a noncovalent association that is critical for transport of the Env complex from the Golgi to the cell surface. The gp120–gp41 interaction is fairly weak, and a substantial amount of gp120 is shed from the surface of Env-expressing cells.
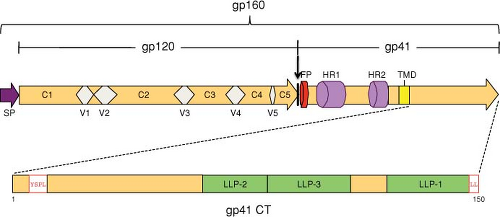 Figure 49.9. Linear representation of the human immunodeficiency virus 1 (HIV-1) Env glycoprotein. The vertical arrow indicates the site of gp160 cleavage to gp120 and gp41; SP denotes the signal peptide. In gp120, variable domains (V1 to V5) and conserved sequences (C1 to C5) are indicated. In the gp41 ectodomain, several domains are indicated: the N-terminal fusion peptide (FP) and the two heptad repeats (HR1 and HR2). The transmembrane domain (TMD) is represented by a yellow box. In the ∼150 amino acid gp41 cytoplasmic tail (CT), the Tyr-Ser-Pro-Leu (YSPL) and Leu-Leu (LL) motifs implicated in Env trafficking and internalization are indicated. The putative helical motifs—or lentiviral lytic peptides (LLP-1, LLP-2, and LLP-3)—are shown. (Adapted from Checkley MA, Luttge BG, Freed EO. HIV-1 envelope glycoprotein biosynthesis, trafficking, and incorporation. J Mol Biol 2011;410:582–608, copyright 2011, with permission from Elsevier.) |
The HIV Env glycoprotein complex, in particular the gp120 component, is very heavily glycosylated; approximately half the molecular mass of gp160 is composed of oligosaccharide side chains. During transport of Env from its site of synthesis in the ER to the plasma membrane, many of the side chains are modified by the addition of complex sugars. The numerous oligosaccharide side chains form a sugar “cloud,” obscuring much of gp120 from host immune recognition. As shown in Figure 49.10, gp120 contains interspersed conserved (C1 to C5) and variable (V1 to V5) domains. The Cys residues present in the gp120 proteins of different isolates are highly conserved and form disulfide bonds that link the first four variable regions in large loops.437
After its arrival at the cell surface, the gp120–gp41 complex is rapidly internalized. Several studies have demonstrated that a Tyr-X-X-Leu sequence in the gp41 cytoplasmic tail (approximately five residues from the membrane-spanning domain) (see Fig. 49.9) is at least partially responsible for this rapid internalization.417,645 Analogous motifs are also present in the TM Env proteins of HIV-2, SIV, and several other retroviruses. Tyr-based motifs are known to mediate endocytosis of cellular plasma membrane proteins by binding the μ2 chain of clathrin-associated adaptor protein-2 (AP-2) complexes, and such interactions have been observed with the HIV-1 and SIV gp41 cytoplasmic domains.69,568 A dileucine motif at the C-terminus of the gp41 cytoplasmic tail also participates in Env internalization and trafficking via interactions with the AP-1 complex52,94,820 (Fig. 49.10). Rapid Env internalization could help HIV-1 evade the host immune response and limit Env-induced cytopathicity. The cytoplasmic tail of HIV-1 and SIV gp41 has also been reported to activate NF-κB, potentially providing a stimulus for T-cell activation, thereby enhancing viral infection.612
CD4 Binding
The first step in HIV/SIV infection involves an interaction between gp120 and CD4, the major cell surface receptor for primate lentiviruses (for review, see77). CD4 is a 55-kD member of the immunoglobulin (Ig) superfamily; it is composed of a highly charged cytoplasmic domain, a single hydrophobic
membrane-spanning domain, and four distinct extracellular domains, D1 to D4.475 CD4 normally functions to stabilize the interaction between the T-cell receptor on the surface of T lymphocytes and class II major histocompatibility complex (MHC-II) molecules on antigen-presenting cells. The high-affinity CD4 binding site for gp120 has been localized to a small segment of the N-terminal extracellular domain, analogous to the second complementarity-determining region (CDR-2) loop of an Ig light chain variable domain. CD4 binding determinants in Env map to the C3 and C4 domains of gp120, although a more discontinuous, conformation-dependent domain is involved in high-affinity gp120–CD4 binding. CD4 binding serves not only to promote virion attachment to the target cell but also to induce conformational changes in gp120 and gp41 that activate Env fusogenicity.661,751 In most HIV-1 isolates, CD4 binding induces conformational changes in gp120 that promote gp120 binding to co-receptors.
membrane-spanning domain, and four distinct extracellular domains, D1 to D4.475 CD4 normally functions to stabilize the interaction between the T-cell receptor on the surface of T lymphocytes and class II major histocompatibility complex (MHC-II) molecules on antigen-presenting cells. The high-affinity CD4 binding site for gp120 has been localized to a small segment of the N-terminal extracellular domain, analogous to the second complementarity-determining region (CDR-2) loop of an Ig light chain variable domain. CD4 binding determinants in Env map to the C3 and C4 domains of gp120, although a more discontinuous, conformation-dependent domain is involved in high-affinity gp120–CD4 binding. CD4 binding serves not only to promote virion attachment to the target cell but also to induce conformational changes in gp120 and gp41 that activate Env fusogenicity.661,751 In most HIV-1 isolates, CD4 binding induces conformational changes in gp120 that promote gp120 binding to co-receptors.
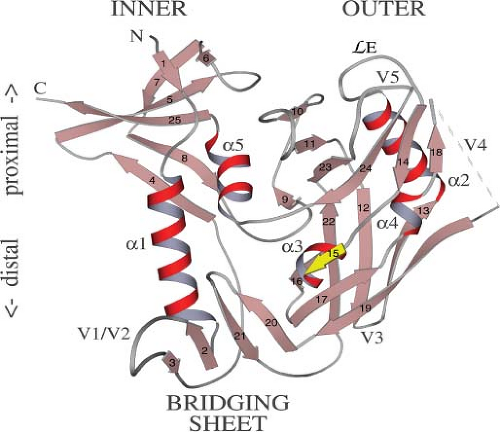 Figure 49.10. Ribbon diagram of the gp120 core. In this orientation, the viral membrane would be at the top, the target cell membrane at the bottom. The inner and outer domains are connected by a four-stranded β-bridging sheet. The remnants of variable loops V1/V2, V3, V4, and V5 are shown. (Reprinted from Kwong PD, Wyatt R, Robinson J, et al. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 1998;393:648–659. Copyright © 1998, Nature.) |
The crystallization of a gp120 “core” domain (an unglycosylated gp120 derivative lacking the V1/V2 and V3 loops and the N- and C-termini) complexed with fragments of CD4 and a neutralizing antibody contributed greatly to our understanding of the gp120–CD4 interaction.416,819 The core structure reveals two major domains (referred to as the “inner” and “outer” domains) connected by a so-called bridging sheet (see Fig. 49.10). The latter is composed of a four-stranded, antiparallel β-sheet derived from sequences in the V1/V2 stem and portions of C4. Comparison of the CD4-bound HIV-1 gp120 structure with that of the non–CD4-bound (unliganded) gp120 from SIVmac reveals that gp120, the inner domain in particular, undergoes a remarkably extensive conformational change upon CD4 binding.118 This shift in conformation leads to the formation of the bridging sheet, which is absent in the unliganded structure.
Co-receptor Interactions
Soon after the identification of CD4 as the major HIV/SIV receptor, it was recognized that this protein is not sufficient for HIV-induced membrane fusion and virus entry. In the mid-1990s, a number of studies demonstrated that members of the G protein–coupled receptor superfamily of seven-transmembrane domain proteins provided the long-sought co-receptor function.14,127,181 As mentioned earlier, the two major co-receptors for HIV/SIV infection are CXCR4 and CCR5. In recognition of the importance of CXCR4 and CCR5 in determining HIV-1 tropism, a nomenclature scheme was devised based on co-receptor usage: viruses (generally M-tropic) that use CCR5 are denoted R5 isolates, strains (generally TCL-tropic) that preferentially use CXCR4 are referred to as X4 viruses, and dual-tropic strains that use both CCR5 and CXCR4 are denoted R5/X4 isolates47 (Table 49.1).
Since the discovery of HIV/SIV co-receptors, numerous additional studies refined our understanding of the activity and function of these proteins. Some of the more important findings are described here; for further information, the reader is referred to more-detailed reviews.48
It is clear that, depending on the virus isolate, multiple sequences in the N-terminal domain and extracellular loops of CCR5 and CXCR4 can influence co-receptor function, and that the gp120–co-receptor interaction is complex.
Certain laboratory isolates of HIV-1, HIV-2, and SIV use co-receptors in a CD4-independent manner, that is, as primary receptors.205,499,613 In fact, the identification of CD4-independent HIV and SIV isolates suggests that the use of chemokine receptors in lentiviral infection may have predated the involvement of CD4.
Direct interactions between co-receptor and gp120 have been detected. These interactions are greatly stimulated by, or are dependent on, the presence of CD4.425,758,816
In addition to CXCR4 and CCR5, a number of other chemokine receptors and related proteins can serve as co-receptors for HIV/SIV fusion and infection in cultured cells. These include CCR2b, CCR3, CCR8, APJ, Bonzo (STRL33), BOB (GPR15), and US28 (for review, see48). What role, if any, these alternative co-receptors play in HIV/SIV infection in vivo is unclear.
Several domains within gp120 directly or indirectly function in Env–co-receptor interactions. Consistent with its influence on HIV-1 tropism, the V3 loop plays a major role. In addition to the involvement of variable regions in co-receptor interaction, highly conserved portions of gp120 also interact with co-receptors. Following the initial report of the gp120 core crystal structure in 1998, a number of additional HIV-1 and SIV gp120 structures have been published. Together, these structures have provided a number of key insights into the gp120 structure/function relationship. As mentioned earlier, gp120 undergoes extensive conformational changes upon binding to CD4. Particularly significant is that the co-receptor binding site is essentially created following CD4 binding.118 As a result, the co-receptor binding site, which would be highly susceptible to neutralizing antibodies, exits only transiently after gp120 has contacted CD4 on the target cell membrane. These structural findings are consistent with earlier reports demonstrating that binding of antibodies to the co-receptor-interacting surface of gp120 is enhanced by CD4.662,751 Interestingly, the co-receptor–binding surface is constitutively exposed in variants selected to replicate in a CD4-independent fashion.322,402 Because their co-receptor–binding surface is constitutively exposed, CD4-independent isolates tend to be hypersensitive to neutralization.
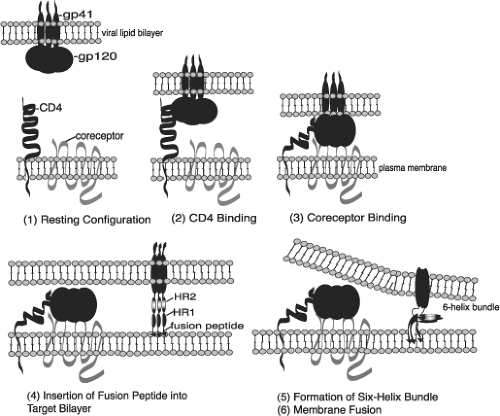 Figure 49.11. Schematic representation of the steps leading to membrane fusion. In the resting configuration, the Env glycoprotein complex is in its native state (1). CD4 binding (2) induces conformation changes in Env that facilitate coreceptor binding (3). After the formation of a ternary gp120–CD4–co-receptor complex, gp41 adopts a hypothetical conformation that allows the fusion peptide to insert into the target lipid bilayer (4). The formation of the gp41 six-helix bundle, which involves antiparallel interactions between the gp41 heptad repeats (HR1 and HR2) (5), brings the viral and cellular membranes together and membrane fusion takes place (6). |
Certain individuals, despite persistent high-risk behavior, remain HIV-1 uninfected. The discovery of CCR5 and CXCR4 as HIV-1 co-receptors raised the possibility that these individuals inherited mutant co-receptor alleles. Indeed, a mutant CCR5 allele, referred to as CCR5/Δ32, contains a 32-bp deletion and encodes a truncated protein that is not efficiently expressed at the cell surface and cannot function as an HIV-1 co-receptor.176,335,457,654 Homozygotes for CCR5/Δ32 are only rarely infected with HIV-1, highlighting the protective benefits of inheriting this allele. Other inherited polymorphisms in co-receptor coding regions or regulatory sequences have been reported to influence HIV-1 transmission or HIV-1–induced disease progression, though the effects are not as clear as with the CCR5/Δ32 allele.
Membrane Fusion
The primary function of viral Env glycoproteins is to promote a membrane fusion reaction between viral and target cell membranes. This membrane fusion event enables the viral core to gain entry into the host cell cytoplasm. The observation that CD4 induces conformational changes in gp120 that enhance interaction with co-receptors suggests that events leading up to membrane fusion occur sequentially (Fig. 49.11). gp120 first interacts with CD4; a ternary CD4–co-receptor–gp120 complex then forms; and finally, conformational changes take place in gp41 that trigger membrane fusion. Although fusion between the viral and cellular membrane is widely believed to occur directly at the plasma membrane after CD4 and co-receptor engagement, it has been reported that fusion can also occur in a low-pH endosomal compartment.527
Mutational analyses demonstrated that a hydrophobic region at the N-terminus of gp41, the so-called fusion peptide, plays a central role in membrane fusion mediated by HIV-1,235 HIV-2,234 and SIV74 (see Fig. 49.9). C-terminal to the gp41 fusion peptide are two amphipathic heptad repeat (HR) domains (see Fig. 49.9) that are also required for membrane fusion. Mutations in the N-terminal HR (HR1) impair infectivity and membrane fusion activity,195 and peptides derived from HR1 and HR2 exhibit potent antiviral activity in culture.802 Structures for the ectodomain of HIV-1 and SIV gp41, including the HRs, were determined by x-ray crystallography and nuclear magnetic resonance (NMR) spectroscopy.95,113,743,795 These studies obtained fundamentally similar trimeric structures in which HR1 and HR2 pack in an antiparallel fashion to generate a six-helix bundle (Figs. 49.11 and 49.12). The HR1s form a coiled-coil in the center of the bundle, with the HR2s packing into hydrophobic grooves on the outside. The gp41 ectodomain structure resembles that of the fusion-competent (low-pH-induced) form of influenza HA2, making it likely that HIV/SIV Env glycoproteins trigger membrane fusion by the same “spring-loaded” mechanism proposed for influenza virus.91 Peptides corresponding to the HRs inhibit fusion (and virus infectivity) by interacting with their complementary binding partner on gp41, thus preventing gp41 itself from forming the six-helix bundle. Binding studies using native HIV-1 Env indicate that gp41 interacts with HR2 peptide only after CD4 binding, suggesting that HR1 and HR2 undergo conformation changes after CD4 binding and that these rearrangements are required for membrane fusion.244 The cytoplasmic domain of gp41 also plays a role in modulating the fusion activity of Env, in part by regulating the levels of cell surface expression of the gp120–gp41 complex and by influencing the conformational of gp120 and the ectodomain of gp41 (for review see117).
 Figure 49.12. Trimeric structure of simian immunodeficiency virus (SIV) gp41 ectodomain residues 27 to 149. The N-terminal heptad repeats (HR1s) are located on the inside of the six-helix bundle; the C-terminal heptad repeats (HR2s) pack into hydrophobic grooves on the outside. The three subunits are shown in green, blue, and red. (Reprinted from Caffrey M, Cai M, Kaufman J, et al. Three-dimensional solution structure of the 44 kDa ectodomain of SIV gp41. EMBO J 1998;17:4572–4584, with permission from Oxford University Press.) |
Whereas gp41 interacts directly with the lipid bilayer to catalyze the membrane fusion reaction, a variety of domains within gp120 are involved in activating Env fusogenicity. The V3 loop of HIV-1 gp120, which elicits isolate-specific neutralizing antibodies, is an essential player in membrane fusion. Mutations throughout the V3 loop of HIV-1236 and the analogous region of HIV-2234 block syncytium formation and virus infectivity. The importance of V3 in membrane fusion appears to be in part a consequence of its role in gp120–co-receptor interactions. The structure of the V3 loop in the context of the CD4-bound gp120 core reveals that V3 protrudes out from the core, toward the target cell membrane and the co-receptor.331
It has become apparent that the lipid composition of the target cell plasma membrane plays an important role in membrane fusion. Of particular interest is the concept that membrane fusion may take place in plasma membrane microdomains known as lipid rafts. These microdomains, which are enriched in glycosphingolipids and cholesterol, appear to be involved in the entry of a number of enveloped viruses, including HIV-1 (for review see784). Depletion of cholesterol from either the virus particle or the target cell membrane disrupts fusion,573 as does depletion of specific glycosphingolipids.88,336 The cholesterol-binding compound amphotericin B methyl ester (AME) is also a very potent inhibitor of HIV-1 infectivity.782 Interestingly, HIV-1 evades AME by acquiring mutations in the cytoplasmic tail of gp41 that cause the tail to be cleaved by PR, thereby allowing infection to proceed even in the presence of the inhibitor.783 In addition to promoting cell-free HIV-1 infection, lipid rafts may play an important role in HIV-1 transmission between T cells366 and in virus uptake into DCs.97
Progress in understanding the molecular mechanism of membrane fusion has led to the development of inhibitors that block virus fusion and entry (for review see752). One of these fusion inhibitors, T-20 (enfuvirtide), is now in clinical use. T-20 is a 36–amino acid, gp41 HR2-derived peptide that inhibits HIV-1 replication both in vitro and in infected patients.393,419 This peptide interacts directly with HR1 during the conformational changes that take place after CD4 binding. The interaction of T-20 with gp41 prevents six-helix-bundle formation, thereby blocking fusion. Resistance to T-20 arises both in vitro and in patients, primarily through mutations in HR1.630 Inhibitors that block gp120 binding to CD4 are being tested, and a number of CCR5 and CXCR4 antagonists are under development. Although CXCR4 knockout mice display a range of severe defects in hematopoiesis, central nervous system development, and gastrointestinal tract vascularization,739,864 no observable negative consequences of absent CCR5 function are evident in individuals homozygous for the CCR5/Δ32 mutation. Furthermore, in 2009, it was reported that a patient was apparently cured of HIV-1 infection following transplantation
of stem cells from a CCR5/Δ32 homozygous individual.341 These observations suggest that inhibitors targeting CCR5 should be well tolerated. One CCR5-based inhibitor, maraviroc, is currently approved for clinical use in patients harboring R5-tropic strains of HIV-1 (for review see583). Maraviroc is thought to function by binding extracellular domains of CCR5 and inducing an allosteric change in CCR5 conformation that prevents gp120 binding.211 Resistance to maraviroc can arise in culture by acquisition of gp120 V3 loop mutations that allow HIV-1 to utilize the drug-bound co-receptor.800 In patients, CCR5 inhibitor resistance typically results from selection for CXCR4 strains that were present at low levels prior to the initiation of therapy.533 In addition to use of small-molecule co-receptor antagonists like maraviroc, strategies aimed at knocking down CCR5 expression in HIV-1–infected individuals are being contemplated. A strategy currently under development involves the use of zinc-finger nucleases that disrupt the CCR5 gene (for review see 102).
of stem cells from a CCR5/Δ32 homozygous individual.341 These observations suggest that inhibitors targeting CCR5 should be well tolerated. One CCR5-based inhibitor, maraviroc, is currently approved for clinical use in patients harboring R5-tropic strains of HIV-1 (for review see583). Maraviroc is thought to function by binding extracellular domains of CCR5 and inducing an allosteric change in CCR5 conformation that prevents gp120 binding.211 Resistance to maraviroc can arise in culture by acquisition of gp120 V3 loop mutations that allow HIV-1 to utilize the drug-bound co-receptor.800 In patients, CCR5 inhibitor resistance typically results from selection for CXCR4 strains that were present at low levels prior to the initiation of therapy.533 In addition to use of small-molecule co-receptor antagonists like maraviroc, strategies aimed at knocking down CCR5 expression in HIV-1–infected individuals are being contemplated. A strategy currently under development involves the use of zinc-finger nucleases that disrupt the CCR5 gene (for review see 102).
Reverse Transcription
By definition, retroviruses possess the ability to convert their single-stranded RNA genomes into double-stranded DNA during the early stages of the infection process.31,747 The enzyme that catalyzes this reaction is RT, in conjunction with its associated ribonuclease H (RNaseH) activity. Retroviral RTs have two enzymatic centers: (a) a DNA polymerase that can copy either RNA templates (for “minus-strand” DNA synthesis) or DNA templates (for second- or “plus-strand” DNA synthesis) and (b) RNaseH (for the degradation of the tRNA primer and genomic RNA present in DNA-RNA hybrid intermediates). RT was the first viral protein targeted by antiretroviral therapy, and RT inhibitors remain central to the treatment of HIV-infected patients (for reviews see337,703).
Uncoating occurs after membrane fusion and entry of the viral nucleoprotein complex into the cytosol, giving rise to the reverse transcription complex (RTC). This multiprotein complex is composed not only of RNA and RT but also CA, NC, IN, and Vpr. While it was initially proposed that most if not all of the CA protein was lost from the RTC during uncoating, a variety of observations, including the fact that CA is the target for host restriction factors that disrupt reverse transcription (see later), support the hypothesis that at least some CA protein is retained in this complex. Recent findings suggest that uncoating and reverse transcription are linked; in the presence of an inhibitor of reverse transcription, uncoating is significantly delayed.338
The retroviral genome is packaged into the virion as two copies of single-stranded RNA. As discussed earlier, the two RNAs are held together in part by the DIS near their 5′ ends (see Fig. 49.7). Although each retroviral particle carries two strands of RNA, it appears that only one provirus is formed per virion.330 Retroviruses are therefore referred to as pseudodiploid. As with most other DNA polymerases, RT is dependent on the 3′-OH group of an RNA or DNA primer to initiate polymerization. Retroviruses use specific tRNAs (tRNALys3 in the case of HIV-1) to initiate DNA synthesis. The mechanism of tRNA selection and placement on the template is complex, involving interactions with RT and NC as well as with the 18-nt pbs near the 5′ end of the viral genome (see Fig. 49.7).
Reverse transcription of the retroviral RNA genome to a double-stranded DNA copy takes place after viral entry into the target cell and proceeds via a series of steps that are outlined briefly here (see e-Fig. 49.2A). This is also described in Figure 47.7 of Chapter 47.
Minus-strand DNA synthesis is initiated from the 3′-OH of the tRNA bound to the pbs. DNA synthesis then proceeds to the 5′ end of the genome.
RNaseH digests the RNA portion of the newly formed RNA-DNA hybrid, freeing the resulting short, single-stranded DNA fragment (known as the minus-strand strong-stop DNA).
The minus-strand strong-stop DNA is transferred to the 3′ end of the genome, where it hybridizes by virtue of a short region of homology (the “repeated” or R region) present at both 5′ and 3′ ends of the RNA genome.
Minus-strand synthesis, accompanied by RNaseH-mediated degradation of the RNA in the resulting RNA-DNA hybrid, continues to the pbs at the 5′ end of the genome.
Fragments of RNA that were not removed by RNaseH serve as primers for plus-strand synthesis. The major site of plus-strand priming is the PPT (see e-Fig. 49.2A, top); however, residual RNA fragments that remain hybridized to regions outside the PPT can also be used for priming plus-strand synthesis. In the case of HIV-1, one such region, known as the central PPT, appears to be particularly important in this regard.114
After plus-strand synthesis copies a portion of the tRNA primer, RNaseH removes the tRNA. This exposes the pbs at the 3′ end of the plus-strand DNA, allowing the plus-strand DNA to hybridize with the homologous region at the 3′ end of the minus-strand DNA (second-strand transfer).
Plus- and minus-strand syntheses proceed to completion. Plus-strand synthesis terminates at the end of the minus strand and, for HIV-1, at a sequence known as the central termination signal (CTS).115 The position of the central PPT upstream of the CTS results in the displacement of approximately 100 nt of plus-strand DNA and the formation of a triplex DNA structure. It has been suggested that this triplex, or DNA flap structure, plays a role in nuclear import of the viral PIC,846 though this has been controversial. The final product of reverse transcription is a double-stranded DNA molecule capable of serving as the substrate for integration.
In addition to providing an essential function in the virus life cycle, the enzymatic activity of RT is routinely used in the laboratory to quantitatively monitor levels of progeny virions present in the supernatant of infected cultures and to elucidate the molecular details of virus replication. Also, the detection of viral DNA postinfection by real-time polymerase chain reaction (PCR) provides one of the most reliable methods to date for detecting entry and postentry steps in the virus replication cycle. Once inside the nucleus, some double-stranded viral DNA is ligated to generate circular forms; although these circular DNA products do not integrate, their formation is often used to monitor nuclear import (see later).
The mature HIV-1 RT holoenzyme is a heterodimer of 66- and 51-kD subunits. The 51-kD subunit (p51) is derived from the 66-kD (p66) subunit or some larger precursor by proteolytic removal of the C-terminal 15-kD fragment of p66 by PR (Fig. 49.6). The structure of HIV-1 RT has been determined by x-ray crystallography in a number of studies. The enzyme has been crystallized in several contexts: (a) unliganded,633 (b) bound to
a short DNA duplex and the Fab portion of an anti-RT antibody,354 (c) covalently linked to a complex of primer/template and deoxyribonucleotide triphosphate (dNTP),332 and (d) bound to an RNA-DNA duplex.655 A number of structures are also available for RT bound to nonnucleoside RT inhibitors (NNRTIs). The crystal structure of HIV-1 RT reveals that the p66 and p51 subunits are folded into similar subdomains, but these subdomains are arranged quite differently in p66 and p51. The p66 subunit can be visualized as a right hand, with the polymerase active site within the palm, and a deep template-binding cleft formed by the palm, fingers, and thumb subdomains355 (Fig. 49.13 and 49.14). The polymerase domain is linked to RNaseH by the connection subdomain. The active site, located in the palm, contains three critical Asp residues (Asp-110, Asp-185, and Asp-186) in close proximity, and two coordinated Mg2+ ions. Mutation of these Asp residues abolishes the polymerase activity of RT. The orientation of the three active-site Asp residues is similar to that observed in other DNA polymerases (e.g., the Klenow fragment of Escherichia coli DNA Pol I).22 The p51 subunit appears to play a structural role and does not form a polymerizing cleft; Asp-110, Asp-185, and Asp-186 of p51 are buried within the subunit. Approximately 18 bp of the primer/template duplex lie in the nucleic acid–binding cleft, stretching from the polymerase to the RNaseH active sites. In the RT–primer/template–dNTP structure,332 the presence of a dideoxynucleotide (ddNTP) at the 3′ end of the primer allows visualization of the catalytic complex blocked just prior to attack on the incoming dNTP. Comparison with previously obtained structures suggests a model whereby the fingers close in to trap the template and dNTP prior to nucleophilic attack of the 3′-OH of the primer on the incoming dNTP. After the addition of the incoming dNTP to the growing chain, the fingers adopt a more open configuration, thereby releasing the pyrophosphate and enabling RT to bind the next dNTP. HIV-1 RNaseH, whose structure has been determined by x-ray crystallography, displays a global folding similar to that of E. coli RNaseH.169 The structure of human RNaseH1 bound to an RNA-DNA hybrid has allowed the modeling of HIV-1 RT RNaseH complexed with its substrate.566
a short DNA duplex and the Fab portion of an anti-RT antibody,354 (c) covalently linked to a complex of primer/template and deoxyribonucleotide triphosphate (dNTP),332 and (d) bound to an RNA-DNA duplex.655 A number of structures are also available for RT bound to nonnucleoside RT inhibitors (NNRTIs). The crystal structure of HIV-1 RT reveals that the p66 and p51 subunits are folded into similar subdomains, but these subdomains are arranged quite differently in p66 and p51. The p66 subunit can be visualized as a right hand, with the polymerase active site within the palm, and a deep template-binding cleft formed by the palm, fingers, and thumb subdomains355 (Fig. 49.13 and 49.14). The polymerase domain is linked to RNaseH by the connection subdomain. The active site, located in the palm, contains three critical Asp residues (Asp-110, Asp-185, and Asp-186) in close proximity, and two coordinated Mg2+ ions. Mutation of these Asp residues abolishes the polymerase activity of RT. The orientation of the three active-site Asp residues is similar to that observed in other DNA polymerases (e.g., the Klenow fragment of Escherichia coli DNA Pol I).22 The p51 subunit appears to play a structural role and does not form a polymerizing cleft; Asp-110, Asp-185, and Asp-186 of p51 are buried within the subunit. Approximately 18 bp of the primer/template duplex lie in the nucleic acid–binding cleft, stretching from the polymerase to the RNaseH active sites. In the RT–primer/template–dNTP structure,332 the presence of a dideoxynucleotide (ddNTP) at the 3′ end of the primer allows visualization of the catalytic complex blocked just prior to attack on the incoming dNTP. Comparison with previously obtained structures suggests a model whereby the fingers close in to trap the template and dNTP prior to nucleophilic attack of the 3′-OH of the primer on the incoming dNTP. After the addition of the incoming dNTP to the growing chain, the fingers adopt a more open configuration, thereby releasing the pyrophosphate and enabling RT to bind the next dNTP. HIV-1 RNaseH, whose structure has been determined by x-ray crystallography, displays a global folding similar to that of E. coli RNaseH.169 The structure of human RNaseH1 bound to an RNA-DNA hybrid has allowed the modeling of HIV-1 RT RNaseH complexed with its substrate.566
 Figure 49.13. Ribbon diagrams of the p66 (left) and p51 (right) reverse transcriptase (RT) subunits. Polymerase active site and fingers, palm, thumb, and connection subdomains are shown. (Modified by Kalyas Das and Eddy Arnold from Jacobo-Molina A, Ding J, Nanni RG, et al. Crystal structure of human immunodeficiency virus type 1 reverse transcriptase complexed with double-stranded DNA at 3.0 Å resolution shows bent DNA. Proc Natl Acad Sci U S A 1993;90:6320–6324.) |
 Figure 49.14. Human immunodeficiency virus 1 (HIV-1) reverse transcriptase (RT) bound to an RNA/DNA template/primer. (Modified by Karen Kirby and Stefan Sarafianos from Singh K, Marchand B, Kirby KA, et al. Structural aspects of drug resistance and inhibition of HIV-1 reverse transcriptase. Viruses 2010;2:606–638.) |
The high rate of variation among HIV-1 populations poses one of the fundamental challenges to effectively controlling this pathogen. Highly variable virus populations are largely a consequence of rapid virus replication rates, coupled with the error-prone nature of RT (which lacks a proofreading function) and frequent template switching during reverse transcription141 (for review see733). Cell-free error rates for purified RTs have been determined,38,616,631 as have in vivo retrovirus mutation rates.2,192,491,585,586 Errors during retrovirus replication are also likely to be introduced by the cellular DNA-dependent RNA Pol II during transcription of the viral RNA. The cell-free error
rate measured for HIV-1 RT is more than 10-fold higher than the mutation rate, suggesting that the in vitro reaction lacks factor(s) that increase fidelity.491 The total HIV-1 in vivo mutation rate (a composite of substitutions, frameshifts, simple deletions, and deletions with insertions) was measured at ∼1 to 3 × 10−5 per cycle of replication.2,491
rate measured for HIV-1 RT is more than 10-fold higher than the mutation rate, suggesting that the in vitro reaction lacks factor(s) that increase fidelity.491 The total HIV-1 in vivo mutation rate (a composite of substitutions, frameshifts, simple deletions, and deletions with insertions) was measured at ∼1 to 3 × 10−5 per cycle of replication.2,491
As discussed earlier, retroviral particles contain two copies of single-stranded RNA. During reverse transcription, RT frequently switches from one template to the other; this occurs on the order of ∼30 times per infection event.34 RT also undergoes intramolecular jumps.584 Intramolecular jumps lead to mutations (e.g., deletions, insertions, and duplications), whereas intermolecular jumps generate recombinants if the two packaged RNAs are not genetically identical.329 The intermolecular template switches that lead to recombination can occur when RT encounters a break in the RNA during polymerization—the so-called forced copy choice model.142 Jumps during minus-strand synthesis can occur in the absence of strand breaks,371 and the stability of RT association with template is influenced by the balance between the enzyme’s RNaseH activity and polymerase activity—the “dynamic copy-choice” model.342 The impact of high rates of recombination and high replication rates on HIV-1 populations is substantial. As noted earlier, a significant percentage (∼20%312) of sequenced virus isolates are intersubtype recombinants, and recombinants constitute the predominant strains currently circulating in certain parts of the world. Recombination also provides a mechanism for the rapid generation of multiple-drug-resistant HIV-1 variants.
RT has long been a target in the search for antiviral compounds. RT inhibitors are now routinely administered to AIDS patients, usually in combination with PR and, more recently, IN inhibitors. There are two major classes of drugs that block reverse transcription: nucleoside RT inhibitors (NRTIs) and NNRTIs. Examples of NRTIs include AZT (zidovudine); 2′-deoxy-3′-thiacytidine (3TC or lamivudine); 2′,3′ dideoxyinosine (ddI or didanosine); and 2′,3′ dideoxycytidine (ddC or zalcitabine). NNRTIs include nevirapine, efavirenz, and delavirdine. NRTIs are nucleotide mimics that lack the 3′-OH and thus act as chain terminators upon incorporation into DNA, whereas the NNRTIs inhibit DNA polymerization by binding a small hydrophobic pocket near the RT active site and inducing a change in the structure of RT that blocks DNA synthesis.401 Resistance to RT inhibitors often develops in patients as viral variants arise encoding mutant forms of RT that are no longer blocked by the inhibitors (for review see656). Resistance to NRTIs generally takes place by one of two mechanisms: (a) resistant RTs acquire the ability to selectively incorporate natural dNTPs but exclude the NRTI, or (b) mutant RTs incorporate the NRTI but subsequently excise it from the terminated primer. Resistance to NNRTIs occurs when mutations in RT interfere with the binding of the drug to the enzyme by disrupting key drug–enzyme interactions, changing the shape of the NNRTI binding pocket, or preventing entry of the drug into the binding pocket.657 Efforts continue to develop new NRTIs that are more difficult for drug-resistant RTs to exclude, and it may ultimately be possible to develop compounds that interfere with NRTI excision. NNRTIs with increased conformational flexibility (“wiggling and jiggling”) may also be more effective and more difficult for HIV-1 to evade if the flexible inhibitor can still bind to the altered pocket in the resistant RTs.162,163 Finally, a significant effort has been directed toward developing inhibitors that target the RNaseH activity of the enzyme.
Postentry Blocks to Lentivirus Infection
TRIM5α and Family Members
While retroviruses have evolved ingenious strategies to usurp host cell machinery to facilitate their replication, they have also been forced to counter host cell defenses aimed at restricting virus infection. Decades ago, it was discovered that cells from mice of specific genetic backgrounds express dominant factors that block infection by certain subtypes of MLV. For example, the Friend virus susceptibility-1 (Fv1) allele452,601 encodes resistance to distinct strains of MLV; Fv1n confers resistance to B-tropic MLV, whereas Fv1b cells cannot be efficiently infected by N-tropic MLV (N-MLV). The viral determinant of N- and B-tropism maps to a specific amino acid (residue 110) in the CA domain of Gag.406 The Fv1 block occurs early postentry, after reverse transcription but before integration.363 Although the mechanism of Fv1 restriction remains a puzzle, it was demonstrated in the mid-1990s that Fv1 encodes an endogenous Gag-like protein.56
Nonmurine cells do not harbor an Fv1 gene.56 However, seemingly analogous postentry restrictions have also been observed in a variety of mammalian cells. N-tropic MLV poorly infects cells from a number of mammalian species.756 HIV-1 infection is inefficient in cells derived from Old World (e.g., African green and rhesus) monkeys, whereas New World (e.g., owl and squirrel) monkey cells are poorly infected by SIVmac.319,323,695 In these cases, infection appears to be blocked at or before reverse transcription.54,150,756 The host factor responsible for postentry restriction of HIV-1 in rhesus macaque cells was demonstrated to be the cytoplasmic body component TRIM5α.727 Expression of rhesus TRIM5α in human cells potently inhibits HIV-1 infection but has no effect on Moloney MLV infectivity. Conversely, knock-down of TRIM5α expression using siRNA in rhesus cells markedly increases HIV-1 infectivity.727 Subsequent studies indicated that human TRIM5α expression is necessary and sufficient to block N-MLV infection in human cells305,383,596,833 and that TRIM5α derived from several New World monkey species restricts infection by SIV from macaques (SIVmac) and African green monkeys (SIVagm) in human cells.713 Rhesus, African green monkey, and human TRIM5α can also diminish infection by the nonprimate lentivirus equine infectious anemia virus (EIAV),305 and cow and rabbit cells express TRIM-family proteins that restrict diverse retroviruses.670,698
As their name implies, TRIM proteins contain three major domains: a RING domain that possesses ubiquitin ligase activity, a B-box 2 domain, and a coiled-coil domain (Fig. 49.15). The α isoform of TRIM5 (TRIM5α) also contains a C-terminal B30.2 or SPRY domain that is absent in other TRIM5 isoforms. This C-terminal B30.2/SPRY domain harbors the determinants responsible for the species specificity of TRIM5α-mediated restriction to retroviral infection553,593,712,729,833; a single amino acid change in the B30.2/SPRY domain of human TRIM5α converts this protein from a weak inhibitor of HIV-1 infection to one that behaves like rhesus TRIM5α in potently restricting HIV-1 infection.833 This specificity is mediated through direct binding between the B30.2/SPRY domain and the CA protein on the incoming RTC.423,450,686 Binding of TRIM5α to CA requires both TRIM5α dimerization and
higher-order multimerization.423,450 The higher-order multimers have been reported to be hexameric,249,559 an observation that is likely relevant to the mechanism of TRIM5 restriction (see later). The importance of TRIM5α in combating retroviral infections is supported by the finding that the CA-binding B30.2/SPRY domain in TRIM5α has undergone positive selection during primate evolution.362,666,712
higher-order multimerization.423,450 The higher-order multimers have been reported to be hexameric,249,559 an observation that is likely relevant to the mechanism of TRIM5 restriction (see later). The importance of TRIM5α in combating retroviral infections is supported by the finding that the CA-binding B30.2/SPRY domain in TRIM5α has undergone positive selection during primate evolution.362,666,712
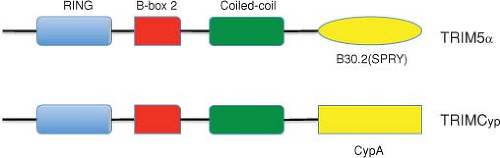 Figure 49.15. Organization of TRIM5α and TRIMCyp. The major domains—RING, B-box 2, coiled-coil, B30.2 (SPRY), and cyclophilin A (CypA)—are shown. Gag-binding determinants of TRIM5α and TRIMCyp reside in the B30.2 (SPRY) and CypA domains, respectively. |
In owl monkey cells, HIV-1 infection is highly inefficient but is significantly enhanced by disrupting the interaction between CA and the cellular protein cyclophilin A (CypA) (see section on Capsid). This observation suggested a link between CA–CypA binding and postentry restriction. The basis for this link was revealed when it was discovered that owl monkey cells express a form of TRIM5α in which the C-terminal B30.2/SPRY domain has been replaced by CypA564,667 (Fig. 49.15). This unusual TRIM5 variant, designated TRIMCyp, restricts HIV-1 infection through a direct binding between the CypA portion of TRIMCyp and CA of the incoming particle. Thus, the ability of TRIMCyp to restrict HIV-1 infectivity in owl monkey cells is eliminated by mutations in the cyclophilin-binding domain of CA or by treating the infected cells with drugs like cyclosporine A that block the CA–cyclophilin interaction.564,667 Interestingly, TRIMCyp has arisen independently in several species of macaque.83,775,806
TRIM family proteins assemble into cytosolic complexes often referred to as “cytoplasmic bodies”.624 Although pre-existing TRIM5α or TRIMCyp cytoplasmic bodies are not required for restriction of retroviral infection,593,711 microscopic examination of cells overexpressing rhesus TRIM5α early after HIV-1 infection revealed a co-localization between TRIM5α-induced cytoplasmic bodies and viral RTCs.100 Several lines of evidence support a model whereby binding of TRIM5α or TRIMCyp to CA on the incoming RTC leads to a destabilization of the RTC and, consequently, a block to reverse transcription: (a) TRIM5α appears to accelerate CA uncoating early postinfection in the context of either HIV-1 or N-MLV.728 (b) Incubation of in vitro–assembled core-like CA complexes with cell lysates containing TRIM5α or TRIMCyp disrupts the assembled structures.65 (c) Treatment of infected cells with proteasome inhibitors reverses the ability of TRIM5α to prevent viral DNA synthesis, suggesting that TRIM5α induces the degradation of RTCs in the proteasome.817 Although proteasome inhibitors rescue the TRIM-induced defect in DNA synthesis, they do not rescue virus infectivity, suggesting a two-step mechanism of action.817 (d) TRIM5α itself undergoes proteasome-dependent degradation after exposure of cells to restriction-sensitive virus.636
Recent structural studies have shed light on the mechanism of action of TRIM5α and TRIMCyp. A rhesus TRIM5α derivative was shown to assemble in vitro into a two-dimensional paracrystalline hexagonal lattice.249 Although assembly could occur spontaneously, it was greatly facilitated by the presence of hexagonal arrays of in vitro–assembled CA. These findings suggest that restricting TRIM proteins assemble on top of incoming CA hexamers, thereby leading to their disassembly and degradation. Consistent with this model, a fragment of rhesus TRIM5α composed of the coiled-coil and B30.2/SPRY domain was shown to bind HIV-1 CA assemblies or virus-derived cores in vitro and induce their disassembly from an extended CA hexameric lattice to individual hexamers.852 Binding of the CA lattice by TRIM5α not only results in CA lattice disassembly and direct antiviral restriction but also contributes to the innate immune response against retroviral infection. Pertel and colleagues597 demonstrated that CA lattice recognition by TRIM5α leads to the generation of free polyubiquitin chains by the ubiquitin-conjugating enzyme UBC13-UEV1A; these polyubiquitin chains in turn activate innate immune signaling through the TAK1 kinase complex.
APOBEC3G and Family Members
While lentiviral gag genes apparently evolved to avoid restriction by TRIM5α and other TRIM family members, another potent suppressor of lentiviral infection drove the evolution of the lentiviral vif (virus infectivity factor) gene. The presence of vif is a highly conserved feature among lentiviruses; a vif product is encoded by all lentiviruses except EIAV. Soon after the discovery of the vif gene,221,706,724 it was noted that the requirement for Vif is strongly producer-cell dependent. Thus, certain cell lines (e.g., 293T, HeLa, COS, SupT1, CEM-SS, and Jurkat) are “permissive” for vif-defective mutants, whereas others, including primary lymphocytes, MDMs, and some T-cell lines, are “nonpermissive.” This observation suggested that a host cell factor in the virus-producing cell plays a role in Vif function. Experiments performed with fusions between permissive and nonpermissive cells indicated the existence of a dominant inhibitory factor whose antiviral activity is countered by Vif.474,702 A major breakthrough came with the discovery that APOBEC3G (apolipoprotein B mRNA-editing enzyme, catalytic polypeptide-like 3G) is the host factor whose antiviral activity is neutralized by Vif. This finding led to a flurry of studies that have at least partially elucidated the mechanism by which APOBEC3G inhibits HIV-1 replication and the molecular basis for the ability of Vif to overcome this inhibition155 (Fig. 49.16). APOBEC3G is a member of a large family of DNA cytidine deaminases that are involved in mRNA editing and immunoglobulin gene diversification.301 These enzymes contain one or two catalytic domains characterized by His/Cys-X-Glu-X23–28-Pro-Cys-X2-Cys motifs. In the absence of Vif, APOBEC3G is incorporated into HIV-1 virions in the producer cell. In the next round of infection, the virion-associated APOBEC3G converts cytidines to uridines during minus-strand DNA synthesis.300,430,487,494,692,849
Cytidine deamination during reverse transcription leads to G-to-A hypermutation of the viral genome, disrupting viral open reading frames. APOBEC3G-induced defects in processing of the tRNA primer, plus-strand transfer during reverse transcription, and DNA integration have also been noted.508 In nonpermissive (e.g., APOBEC3G-expressing) producer cells, Vif induces the degradation of APOBEC3G, thereby counteracting its antiviral activity838 (for review see155). The action of cellular deaminases may contribute to the prevalence of G-to-A mutations (first noted in spleen necrosis virus586 and observed in some HIV-1 sequences in vivo357,771). Some of the antiviral activity of APOBEC3G has been reported to be independent of deamination activity of the protein, particularly at high expression levels.481 While hypermutation is clearly disadvantageous to HIV-1 replication, low levels of APOBEC-induced mutation may contribute to retroviral variation and as such could be beneficial to the virus.651
Cytidine deamination during reverse transcription leads to G-to-A hypermutation of the viral genome, disrupting viral open reading frames. APOBEC3G-induced defects in processing of the tRNA primer, plus-strand transfer during reverse transcription, and DNA integration have also been noted.508 In nonpermissive (e.g., APOBEC3G-expressing) producer cells, Vif induces the degradation of APOBEC3G, thereby counteracting its antiviral activity838 (for review see155). The action of cellular deaminases may contribute to the prevalence of G-to-A mutations (first noted in spleen necrosis virus586 and observed in some HIV-1 sequences in vivo357,771). Some of the antiviral activity of APOBEC3G has been reported to be independent of deamination activity of the protein, particularly at high expression levels.481 While hypermutation is clearly disadvantageous to HIV-1 replication, low levels of APOBEC-induced mutation may contribute to retroviral variation and as such could be beneficial to the virus.651
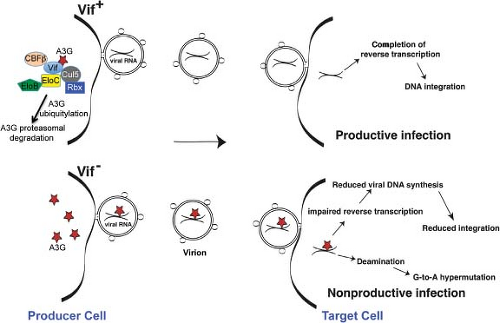 Figure 49.16. Proposed model for inactivation of APOBEC3G by Vif. Top: In nonpermissive (APOBEC3G-expressing) producer cells, Vif links APOBEC3G to the cellular proteins Cul5, elongins B and C, and ring-box-1 (Rbx1) to form an Skp1-cullin F-box (SCF)-like E3 ubiquitin ligase complex, thereby inducing the proteasomal degradation of APOBEC3G and limiting its incorporation into virions. Bottom: In the absence of Vif, APOBEC3G is incorporated into virions. Its presence during reverse transcription in the target cell leads to impaired reverse transcription and integration and deamination of cytidines to uridines, resulting in G-to-A hypermutation. (Adapted from Freed EO. HIV-1 and the host cell: an intimate association. Trends Microbiol 2004;12:170–177.) |
Vif induces the degradation of APOBEC3G through the ubiquitin-proteasome pathway by interacting with the cellular proteins Cul5, elongins B and C, and ring-box-1 (Rbx1) to form an Skp1-cullin F-box (SCF)-like E3 ubiquitin ligase complex.517,838 The transcription co-factor CBF-β is also recruited to this ubiquitin ligase complex356 (Fig. 49.16). The formation of this complex leads to the ubiquitination of APOBEC3G and its degradation in the 26S proteasome. The ability of Vif to induce the degradation of APOBEC3G is species specific; for example, HIV-1 Vif is inactive against simian APOBEC3G, and SIVagm Vif is unable to block the antiviral action of human APOBEC3G. The key difference between human and simian APOBEC3G with respect to degradation by HIV-1 Vif maps to a single residue difference between the two APOBEC3G proteins.71,488,676,822 Murine APOBEC3 is also readily incorporated into HIV-1 virions even in the presence of Vif, making this rodent homolog a potent inhibitor of HIV-1 replication.63,494
The ability of APOBEC3G to restrict HIV-1 replication is highly dependent on its incorporation into virions. APOBEC3G incorporation is rather promiscuous: the human protein can be packaged into, and inhibit, diverse retroviruses, including SIVs, EIAV, and MLV.245,300,487,494 Because these viruses are highly divergent at the protein sequence level, APOBEC3G likely gains access to virions through a highly conserved structural element or by binding to molecules present in all retroviral particles (e.g., RNA). Considering that APOBEC3G acts on the viral nucleic acid during reverse transcription, it presumably must associate with the viral core391 and ultimately with the RTC. Finally, it is reasonable to assume that APOBEC3G is incorporated through interaction with a viral element that is essential for replication; otherwise, HIV-1 could readily escape APOBEC3G restriction simply by mutating the APOBEC3G binding site without the need for acquiring a novel protein (Vif) to counter APOBEC3G activity. Several studies have focused on the mechanism by which APOBEC3G is packaged into particles, in some cases with discordant results. Some investigators have observed a direct binding between APOBEC3G and Gag, in particular the N-terminal portion of NC.11,111,471,669,845 Using a highly sensitive enzymatic assay to measure APOBEC3G levels in virions, others have not observed a requirement for NC in APOBEC3G incorporation.734 Taken together, it appears that although genomic RNA is not absolutely required,111,471,734,845
RNA (viral or cellular)—perhaps in a complex with NC—performs a central function in APOBEC3G packaging. Because RNA encapsidation plays critical roles in particle assembly and virus replication, an RNA-mediated mode of incorporation would be difficult for HIV-1 to escape. RNA-mediated incorporation would also explain the ability of APOBEC3G to gain entry into highly divergent retroviruses.
RNA (viral or cellular)—perhaps in a complex with NC—performs a central function in APOBEC3G packaging. Because RNA encapsidation plays critical roles in particle assembly and virus replication, an RNA-mediated mode of incorporation would be difficult for HIV-1 to escape. RNA-mediated incorporation would also explain the ability of APOBEC3G to gain entry into highly divergent retroviruses.
Humans encode 11 APOBEC family members, with the APOBEC3 genes (APOBEC3A to H) being the most numerous.301 Although APOBEC3G was originally identified as the cellular restriction factor that is inactivated by Vif,692 other APOBECs also display antiviral properties. Most notably, APOBEC3F inhibits HIV-1 replication, and, like APOBEC3G, its activity is suppressed by Vif.63,451,456,801,854 It thus appears that Vif evolved to counter both APOBEC3G and APOBEC3F. APOBEC3B also inhibits HIV-1 infectivity, but, unlike APOBEC3G and APOBEC3F, it is resistant to degradation by Vif.63,188 The biological implications of APOBEC3B’s antiviral activity are unclear, because this APOBEC family member may not be expressed in most primary human tissues.63,188
Some Vif-negative retroviruses have evolved distinct mechanism(s) for evading restriction by cellular cytidine deaminase. HTLV-1 appears to counter APOBEC3G restriction through a C-terminal extension of its NC domain that blocks packaging of APOBEC3G into particles.183 Although MLV infection is inhibited by human APOBEC3G, it is insensitive to all other human APOBEC family members and is resistant to murine APOBEC3 in vitro.63,189 However, in vivo, disruption of the murine APOBEC3 gene leads to enhanced MLV replication and MLV-induced tumor induction.463 Knockout of murine APOBEC3 also increases the susceptibility of mice to mouse mammary tumor virus infection.570 Horses encode six APOBEC3 proteins, one of which is capable of inhibiting HIV-1 replication. Despite efficient packaging into virions, this APOBEC3 does not inhibit EIAV infectivity, suggesting a mechanism of APOBEC evasion that does not rely on virion exclusion.70
The interplay between Vif and APOBEC cytidine deaminases offers novel possibilities for antiretroviral therapy. For example, disrupting the binding between Vif and APOBECs with small-molecule inhibitors would activate the antiviral activity of the cellular enzymes. Interfering with Vif-mediated APOBEC degradation could also theoretically be achieved with proteasome inhibitors (which also block virus budding), by functional disruption of the ubiquitin-conjugating enzymes or ubiquitin ligases specifically responsible for APOBEC ubiquitination, or by blocking the interaction of APOBECs with the SCF-like E3 ubiquitin ligase complex. Some initial progress toward targeting APOBEC and Vif has been made; inhibitors that prevent Vif-mediated APOBEC3G degradation112 or that induce the degradation of Vif itself555 have been reported.
SAMHD1
As mentioned previously, recent studies have demonstrated that the host protein SAMHD1 restricts an early step in lentiviral infection in myeloid cells and is counteracted by the accessory protein Vpx. This topic will be discussed in more detail later in the section on Vpx.
Nuclear Import
Most retroviral genomes gain access to host cell chromosomal DNA during progression of the target cell through the cell cycle.444,634 These retroviruses are therefore unable to efficiently infect noncycling cells. In contrast, lentiviruses, including HIV-1, are able to productively infect nondividing cells (e.g., macrophages). During the events leading to its integration in the nucleus, the newly synthesized viral DNA remains associated with the PIC. This complex, which is composed of both viral and cellular proteins,79,87 must presumably possess the ability to cross the nuclear membrane during interphase to access the host cell genome. Because the HIV-1 PIC—estimated to be ∼56 nm in diameter523—is much larger than the passive diffusion limit for nuclear pores (∼9 nm),505 it has long been assumed that the PIC must harbor determinants that promote active nuclear import. Initially, it was proposed that MA contains the determinants of PIC nuclear import90,777; however, this model was not validated.226,230,232 Subsequent findings implicated Vpr in nuclear import (see section on Vpr). Numerous studies have observed that IN localizes to the nucleus, giving rise to the hypothesis that IN regulates PIC transfer across the nuclear membrane (e.g.,78). This model has also been controversial, because mutations that disrupt the nuclear import of IN can cause defects in multiple aspects of IN function.347,467 It has also been proposed that a cis-acting, centrally located, triple-stranded DNA structure described earlier and known as the central DNA flap, which is a product of lentiviral reverse transcription (see section on Reverse Transcription), promotes nuclear import of the PIC.846 Although inclusion of the central flap improves the infectivity of lentiviral vectors, there is some disagreement as to whether this sequence is required in the context of replication-competent, full-length HIV-1 molecular clones.19,197,454 An HIV-1 chimera-containing MLV IN and lacking all other proposed determinants of nuclear import (e.g., MA, Vpr, and the central DNA flap) retains the ability to infect nondividing cells,825 indicating that none of these elements is critical for HIV-1 nuclear import.
Although the viral determinants of nuclear import, and the mechanism by which they function, remain to be clarified, there is compelling evidence for a connection between CA and nuclear import. It was initially believed that HIV-1 CA is lost from the reverse transcription complex during uncoating; it now appears, however, that a significant amount of CA remains associated with this complex.21,187,512 Analysis of chimeras between MLV (which is unable to infect nondividing cells) and HIV-1 (which can infect nondividing cells) revealed that the ability to infect nondividing cells maps to CA.823 Furthermore, mutations in CA can impose on HIV-1 infection a requirement for cell cycle progression826 and can inhibit nuclear import.187
As mentioned earlier, several genome-wide RNA interference (RNAi) screens have identified a large number of host factors that are putatively required for HIV-1 infection.82,404,857 Among these are nuclear pore proteins and karyopherins—including transportin 3 (TNPO3; also known as TRN-SR2), NUP153, and NUP358/RanBP2—that could play a role in PIC transfer across the nuclear pore. TNPO3 was shown to bind HIV-1 IN and promote PIC nuclear import.131 Subsequent work410 confirmed a role for TNPO3 in HIV-1 infection and demonstrated that most lentiviruses showed a requirement for TNPO3 (feline immunodeficiency virus [FIV] being the exception) but that other retroviruses (e.g., RSV and MLV) did not. This study, however, failed to observe a role for IN–TNPO3 binding in the ability of TNPO3 to promote infection. Rather, the use of the HIV-1/MLV chimeras mentioned earlier
pointed to a role for CA in determining TNPO3 dependency.410 NUP358/RanBP2 bears a cyclophilin domain that has been reported to bind directly to CA; this interaction promotes PIC nuclear import and also influences integration site preference.671
pointed to a role for CA in determining TNPO3 dependency.410 NUP358/RanBP2 bears a cyclophilin domain that has been reported to bind directly to CA; this interaction promotes PIC nuclear import and also influences integration site preference.671
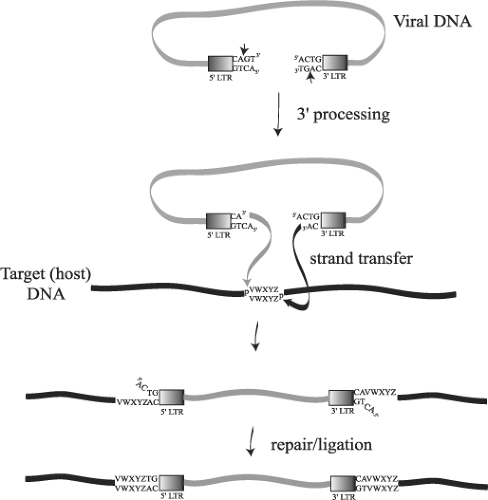 Figure 49.17. Schematic depiction of the integration process. Details are provided in the text. |
In a screen designed to identify dominant factors that interfere with HIV-1 infection, a C-terminally truncated form of cleavage and polyadenylation factor 6 (CPSF6) was shown to block an early step in the HIV-1 infection process—specifically nuclear import—but had no effect on MLV infectivity.432 Selection experiments gave rise to a resistant mutant that, intriguingly, contained a single amino acid change in CA (N74D). This N74D CA mutant was not only resistant to inhibition by the truncated CPSF6 but also displayed an altered requirement for karyopherins and nuclear pore factors. For example, wild-type (WT) HIV-1 infection was inhibited by depletion of TNPO3, RanBP2, and NUP153, but infectivity of the N74D mutant was relatively unaffected.432,504 Conversely, N74D infection was more sensitive than WT to depletion of nuclear pore proteins NUP85 and NUP155. These data again support a role for CA in PIC nuclear import and demonstrate that CA regulates HIV’s utilization of nuclear transport and pore components. CPSF6 is typically localized to the nucleus and overexpression of the full-length, nuclear form has little effect on HIV-1 infection.432 The truncated form that inhibits HIV-1 infection lacks the CPSF6 nuclear localization signal (NLS) and is thus localized to the cytosol. Thus, the shift in CPSF6 localization from nucleus to cytosol may play a key role in its inhibitory activity. Consistent with this model, CPSF6 becomes cytosolic in TNPO3-depleted cells.433 Kewalramani433 showed that in TNPO3-depleted cells CPSF6 becomes cytosolic. Furthermore, CPSF6 depletion reverses the inhibitory effect of TNPO3 depletion. These data suggest that the effect of TNPO3 on HIV-1 is mediated through CPSF6. Because CPSF6 interacts with HIV-1 CA, it is conceivable that this protein plays a regulatory role in HIV-1 infection, potentially as a PIC uncoating or transport factor.
Integration
A distinguishing feature of retrovirus replication is the insertion of a DNA copy of the viral genome into the host cell chromosome after reverse transcription. The integrated viral DNA (the provirus) serves as the template for the synthesis of viral RNAs and is maintained as part of the host cell genome for the lifetime of the infected cell. Retroviral mutants deficient in the ability to integrate generally fail to establish a productive infection.
The integration of viral DNA is catalyzed by IN, a 32-kD protein generated by PR-mediated cleavage of the C-terminal portion of the HIV Gag-Pol polyprotein (see Fig. 49.6). The steps in the integration process were originally elucidated in studies using MLV,87,242 but these findings apply to HIV as well. In all retroviral systems, integration proceeds in the same series of steps (Fig. 49.17):
3′ processing: After its assembly with the viral DNA, IN cleaves immediately downstream from the invariant nucleotide sequence CA, typically removing two nucleotides
from the 3′ termini of both strands of full-length, linear viral DNA, generating a preintegration substrate with 3′ recessed ends.
DNA strand transfer: In the nucleus, IN catalyzes a staggered cleavage in the cellular target DNA. The 3′ recessed ends of viral DNA are joined to the 5′ “overhanging” termini of the cleaved cellular DNA.
Gap repair: The cellular repair machinery fills the gap, thereby completing the integration process.87,242,644 All integrated proviruses terminate with dinucleotides 5′-TG and CA-3′, whereas different genera yield different-sized flanking duplications based on the spacing of the staggered chromosomal DNA cut; the lentiviral duplication is 5 bp. In addition to the linear, double-stranded viral DNA that serves as the substrate for integration, two types of circular DNA are formed in the nucleus: 1-LTR circles and 2-LTR circles. Although these circular DNAs are not substrates for integration, they provide useful markers for nuclear import of the viral DNA, as their formation requires enzymatic machinery present predominantly in the nucleus.
Our understanding of the chemistry of the retroviral integration reaction was greatly assisted by the development of in vitro integration assays. Purified IN, expressed in E. coli, can carry out 3′ processing and strand transfer reactions when combined with short synthetic oligonucleotides that mimic the viral DNA ends and a divalent metal ion (Mg2+ or Mn2+).151,379 Using such systems, IN also catalyzes a reaction known as disintegration, which in essence is the strand transfer reaction in reverse.128 While disintegration may not reflect a physiologically relevant activity of IN, it nevertheless has been useful in understanding the biochemistry of integration. Initial studies observed that the predominant product in in vitro assays using purified HIV-1 IN was a single end joined to one strand of the target, rather than the more physiologically relevant product in which both ends are integrated into the target (concerted or full-site integration).
 Figure 49.18. Schematic representation of an integrase (IN) monomer. The three major structural domains—the N-terminal domain (NTD), catalytic core domain (CCD), and C-terminal domain (CTD)—are shown. The catalytic triad Asp-64, Asp-116, and Glu-152 is represented as D-D-E. |
Retroviral IN proteins are composed of three structurally and functionally distinct domains: an N-terminal zinc-binding domain (NTD), a catalytic core domain (CCD), and a C-terminal domain (CTD) (Fig. 49.18). Problems with low solubility and propensity to aggregate hindered progress in defining the structure of the IN holoenzyme in its active (tetrameric) state. However, the structures of all three domains independently and in various combinations were solved by x-ray crystallography or NMR methods. The NMR structure of the NTD96 revealed three helical bundles with a zinc coordinated by invariant amino acids His-12, His-16, Cys-40, and Cys-43. The structure of the NTD is reminiscent of helical DNA-binding proteins that contain a helix-turn-helix motif. Initially, poor solubility hampered efforts to solve the structure of the CCD. However, attempts at crystallography were successful when a mutation was identified that greatly increased solubility without disrupting in vitro catalytic activity.198 Each monomer of the core domain is composed of a five-stranded β-sheet flanked by helices; this structure bears striking resemblance to that of other polynucleotidyl transferases including RNaseH and the bacteriophage MuA transposase.198,626 Three highly conserved residues are found in analogous positions at the catalytic center of these enzymes; in HIV-1 IN, these residues are Asp-64, Asp-116, and Glu-152—often referred to as the D,D-35-E motif (Fig. 49.18). Mutations in this catalytic triad block HIV-1 IN function both in vivo and in vitro. The IN CTD, whose structure was initially solved by NMR,200,461 adopts a five-stranded β-barrel folding topology reminiscent of Src homology 3 (SH3) domains. The crystal structure of an HIV-1 NTD-CCD IN fragment789 revealed a dimer of dimers, with two inner monomers and two outer monomers, with an extensive intermolecular NTD-CTD interface that provided a preview of the interface evident in the structure of the full retroviral tetramer.
A breakthrough in IN structural biology took place in 2010 with the realization that the IN of prototype foamy virus (PFV) is highly soluble and capable of concerted integration in vitro with short DNA substrates.763 Although divergent at the overall amino acid sequence level, HIV-1 and PFV IN are structurally conserved. The IN tetramer bound to DNA oligonucleotides that mimic the viral DNA ends—which together form the active nucleoprotein complex referred to as the “intasome”—was crystallized and the structure solved.294 This structure revealed that two monomers in the tetramer interact at a dimer interface and are responsible for all contacts with the DNA substrate; the other two monomers are on the outside of the complex and do not interact with each other or with DNA (Fig. 49.19). The residues of the catalytic triad (D,D-35-E) of the inner monomers are oriented close to the 3′-OH of the viral DNA ends. Two metal ions are coordinated at the active site by the residues of the catalytic triad. The addition of model target DNA to the intasome allowed for crystallization of a complex containing both viral and target DNAs.477 In the presence of Mg2+ the strand transfer reaction took place during crystallization, whereas in the absence of divalent metal ions strand transfer was blocked. This allowed capture of postcatalytic strand transfer and prestrand transfer complexes. These structures revealed that the target DNA, which binds in a cleft between the two halves of the intasome complex, adopts a highly bent conformation, allowing the well-separated intasome active sites to access the scissile phosphodiester bonds in the target DNA (Fig. 49.19). The viral (donor) DNA duplex is unpaired at the active site. Conformational changes that occur during catalysis are predicted to render the strand transfer reaction irreversible.477 Following the original publication of the structures described earlier,294,477 a number of additional PFV structures that contained potent inhibitors were solved (discussed later; for review see449); together these reports allowed the development of a structural model for HIV-1 integration that was validated by extensive mutational analysis.409
Although the basic mechanism by which retroviruses integrate their DNAs into the host cell genome is highly conserved, different retroviruses display clear differences in the selection of a cellular target site.525 For example, HIV-1 prefers to
integrate in actively transcribed genes. MLV favors transcription start sites but only weakly selects active genes. ASLV displays a strong preference for neither active genes nor transcription start sites; indeed, for ASLV, high-level transcription appears to be inhibitory for integration.507 Target site selectivity, which impacts upon the development of retroviruses as vehicles for gene therapy, is likely influenced by interactions between IN or other components of the PIC and chromatin-associated host factors. Several cellular proteins have been studied extensively for their contribution to integration and target site selection. The most important among these is a 75-kD, predominantly nuclear, chromatin-associated protein known as LEDGF/p75. A direct interaction between HIV-1 IN and LEDGF/p75 was reported,124,204 and this protein was found to stimulate the strand transfer step of the integration reaction in vitro124 and increase binding of IN to DNA.93 However, early studies were discordant in terms of the effect of LEDGF/p75 depletion on HIV-1 replication.459,769 It soon became apparent that low levels of chromatin-bound LEDGF/p75 were sufficient to promote integration in cells. Near-complete knock-down of LEDGF/p75 levels458 or use of a murine LEDGF/p75 knockout cell line697 revealed markedly reduced (but not fully abolished) HIV-1 infectivity in the absence of detectable LEDGF/p75 expression. Infectivity was also severely compromised by overexpression of the IN-binding domain (IBD) of LEDGF/p75.174,458 In the case of both LEDGF/p75 depletion and LEDGF/p75-IBD overexpression, infectivity was disrupted at the integration step, and the effect was specific for lentiviruses, establishing LEDGF/p75 as a cellular co-factor for lentiviral integration. Studies in which siRNA-resistant LEDGF/p75 mutants were added back to LEDGF/p75-depleted cells identified domains of LEDGF/p75 required for its activity as an integration co-factor.458,697,770 The observation that the LEDGF/p75-IBD and the chromatin-binding regions (the PWWP domain and AT-hook) (Fig. 49.20) were required for function supported the hypothesis that LEDGF/p75 functions to tether the PIC to target chromosomal DNA. The crystal structure of the IN-CCD and LEDGF/p75-IBD complex provided further details of the IN–LEDGF/p75 interaction.123 In addition to a clear role for LEDGF/p75 in directing HIV-1 integration to transcription units, TNPO3 and the nuclear pore protein RanBP2/Nup358 have also been implicated in the targeting of HIV-1 integration. Ocwieja and colleagues567 reported that depleting these genes shifted HIV-1 integration out of gene-dense regions. In contrast, TNPO3 depletion had no effect on the targeting of MLV integration. Use of the aforementioned HIV/MLV chimeras once again mapped the requirement for TNPO3 to Gag.567
integrate in actively transcribed genes. MLV favors transcription start sites but only weakly selects active genes. ASLV displays a strong preference for neither active genes nor transcription start sites; indeed, for ASLV, high-level transcription appears to be inhibitory for integration.507 Target site selectivity, which impacts upon the development of retroviruses as vehicles for gene therapy, is likely influenced by interactions between IN or other components of the PIC and chromatin-associated host factors. Several cellular proteins have been studied extensively for their contribution to integration and target site selection. The most important among these is a 75-kD, predominantly nuclear, chromatin-associated protein known as LEDGF/p75. A direct interaction between HIV-1 IN and LEDGF/p75 was reported,124,204 and this protein was found to stimulate the strand transfer step of the integration reaction in vitro124 and increase binding of IN to DNA.93 However, early studies were discordant in terms of the effect of LEDGF/p75 depletion on HIV-1 replication.459,769 It soon became apparent that low levels of chromatin-bound LEDGF/p75 were sufficient to promote integration in cells. Near-complete knock-down of LEDGF/p75 levels458 or use of a murine LEDGF/p75 knockout cell line697 revealed markedly reduced (but not fully abolished) HIV-1 infectivity in the absence of detectable LEDGF/p75 expression. Infectivity was also severely compromised by overexpression of the IN-binding domain (IBD) of LEDGF/p75.174,458 In the case of both LEDGF/p75 depletion and LEDGF/p75-IBD overexpression, infectivity was disrupted at the integration step, and the effect was specific for lentiviruses, establishing LEDGF/p75 as a cellular co-factor for lentiviral integration. Studies in which siRNA-resistant LEDGF/p75 mutants were added back to LEDGF/p75-depleted cells identified domains of LEDGF/p75 required for its activity as an integration co-factor.458,697,770 The observation that the LEDGF/p75-IBD and the chromatin-binding regions (the PWWP domain and AT-hook) (Fig. 49.20) were required for function supported the hypothesis that LEDGF/p75 functions to tether the PIC to target chromosomal DNA. The crystal structure of the IN-CCD and LEDGF/p75-IBD complex provided further details of the IN–LEDGF/p75 interaction.123 In addition to a clear role for LEDGF/p75 in directing HIV-1 integration to transcription units, TNPO3 and the nuclear pore protein RanBP2/Nup358 have also been implicated in the targeting of HIV-1 integration. Ocwieja and colleagues567 reported that depleting these genes shifted HIV-1 integration out of gene-dense regions. In contrast, TNPO3 depletion had no effect on the targeting of MLV integration. Use of the aforementioned HIV/MLV chimeras once again mapped the requirement for TNPO3 to Gag.567
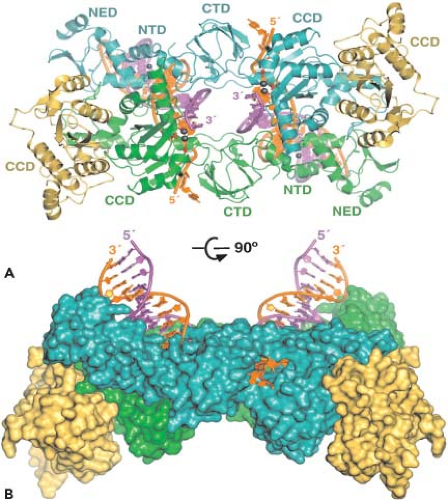 Figure 49.19. Structure of the prototype foamy virus integrase (IN) tetramer in complex with viral DNA ends. The lower image is rotated 90 degrees relative to the upper image. The outer IN chains are yellow; the inner chains are green and cyan. The reactive and nontransferred strands of the viral DNA ends are magenta and orange, respectively. Active-site carboxylates are represented as red sticks. Mn2+ and Zn2+ ions are shown as large and small gray spheres, respectively. The major IN domains (see Fig. 49.18 legend) are labeled. Note that prototype foamy virus also encodes an N-terminal extension domain (NED) not present in human immunodeficiency virus 1 (HIV-1) IN. (Reprinted from Cherepanov P, Maertens GN, Hare S. Structural insights into the retroviral DNA integration apparatus. Curr Opin Struct Biol 2011;21:249–256, copyright 2011, with permission from Elsevier.) |
One of the notable findings to emerge from the LEDGF/p75 depletion studies is that in the absence of LEDGF/p75 expression, HIV’s preference for integrating into transcriptional units is lost.138,697 These results are consistent with the hypothesis that the ability of LEDGF/p75 to tether lentiviral PICs to chromatin is a major determinant for target-site selection. This hypothesis in turn suggests that the targeting of lentiviral integration could be shifted out of transcription units by substituting the chromatin-binding sequences of LEDGF/p75 with chromatin-binding domains from other proteins. Indeed, the infectivity defect conferred by LEDGF/p75 depletion can be rescued with a chimeric protein bearing the LEDGF/p75 IBD
and foreign chromatin-binding domains,515 and such chimeric PIC-tethering factors are able to shift HIV-1 integration target site specificity away from actively transcribed genes.217,261,700 The ability to engineer such custom-designed PIC–chromatin tethering factors and control integration site specificity may have applications in lentiviral-based gene therapy. The function of LEDGF/p75 in promoting HIV-1 integration raises the possibility that the IN–LEDGF/p75 interaction could serve as a target for developing novel antiviral therapeutics. By using the IN-CCD–LEDGF/p75-IBD co-crystal structure in an in silico screen, Debyser and colleagues132 identified small-molecule “LEDGINs” (LEDGF/p75-IN inhibitors) that block the protein interaction in vitro and HIV-1 infection at the integration step.
and foreign chromatin-binding domains,515 and such chimeric PIC-tethering factors are able to shift HIV-1 integration target site specificity away from actively transcribed genes.217,261,700 The ability to engineer such custom-designed PIC–chromatin tethering factors and control integration site specificity may have applications in lentiviral-based gene therapy. The function of LEDGF/p75 in promoting HIV-1 integration raises the possibility that the IN–LEDGF/p75 interaction could serve as a target for developing novel antiviral therapeutics. By using the IN-CCD–LEDGF/p75-IBD co-crystal structure in an in silico screen, Debyser and colleagues132 identified small-molecule “LEDGINs” (LEDGF/p75-IN inhibitors) that block the protein interaction in vitro and HIV-1 infection at the integration step.
 Figure 49.20. Model for integrase (IN)/lens epithelium-derived growth factor (LEDGF) binding to chromosomal DNA. The preintegration complex, containing an IN tetramer (yellow) and viral DNA (vDNA), is depicted in green. The IN-binding domain (IBD) of LEDGF is shown bound to the IN tetramer. The nuclear localization signal (NLS) and AT hooks (AT) are shown in complex with DNA; the PWWP domain is depicted bound to histone proteins. (Reprinted from Adamson CS, Freed EO. Novel approaches to inhibiting HIV-1 replication. Antiviral Res 2010;85:119–141, copyright 2010, with permission from Elsevier.) |
Early studies showed that several classes of agents inhibit IN function in vitro; these included polyanions, nucleotide analogs, and DNA-binding compounds.209 Most of these agents are fairly nonspecific in their mode of action, and many compounds that block activity of purified IN are not inhibitory in assays using purified PICs.210 However, di-keto acid inhibitors were found to be specific and potent inhibitors of the IN-mediated strand transfer reaction both in vitro and in cell-based assays.307 These IN strand transfer inhibitors (INSTIs) bind IN only in the presence of divalent metal and viral DNA and prevent the complex from associating with cellular (target) DNA. Although structurally diverse, INSTIs appear to have common features, including (a) a hydrophobic moiety composed of halobenzyl groups and (b) co-planar oxygen atoms presumed to chelate the metal ions in the active site.509 Work in the early 2000s showed that naphthyridine carboxamides inhibit the replication of an SHIV in rhesus macaques, setting the stage for in vivo use of INSTIs.308 Further structure-activity relationship studies led to the development of raltegravir, also known as MK-0518 or Isentress (Merck Research Laboratories),731 and elvitegravir (Gilead Sciences and Japan Tobacco).659 Raltegravir was approved for clinical use in HIV-infected patients by the U.S. Food and Drug Administration (FDA) in 2007 and elvitegravir was approved in 2012 (reviewed in509).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


