CHAPTER 38 Hormonal Regulation of Energy Metabolism
This chapter considers the role of hormones in maintaining a constant supply of energy to cells in the body during the digestive and interdigestive periods and during fasting and exercise.
OVERVIEW OF ENERGY METABOLISM
Adenosine Triphosphate
Cells continually perform work to maintain their integrity and internal environment, respond to stimuli, and perform their differentiated functions (Fig. 38-1). The absolute minimal amount of energy expenditure is called the basal metabolic rate (BMR) or resting metabolic rate (RMR).
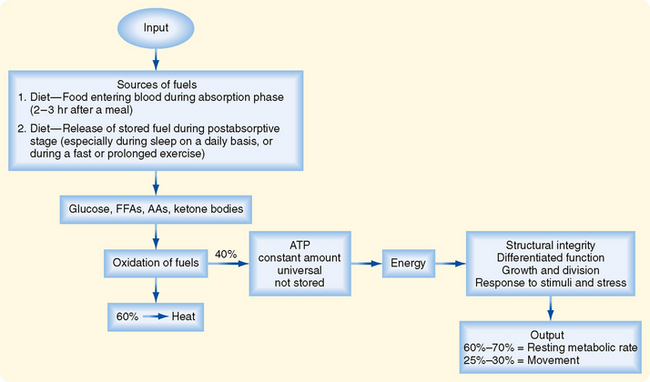
In an adult, other forms of energy expenditure involve
Table 38-1 Estimates of Energy Expenditure in Adults
| Activity | Caloric Expenditure (kcal/min) |
|---|---|
| Basal | 1.1 |
| Sitting | 1.8 |
| Walking, 2.5 miles/hr | 4.3 |
| Walking, 4.0 miles/hr | 8.2 |
| Climbing stairs | 9.0 |
| Swimming | 10.9 |
| Bicycling, 13 miles/hr | 11.1 |
| Household domestic work | 2-4.5 |
| Factory work | 2-6 |
| Farming | 4-6 |
| Building trades | 4-9 |
Data from Kottke FJ. In Altman PL (ed): Metabolism. Bethesda, MD, Federation of American Society for Experimental Biology, 1968.
Cells derive their energy to perform this work primarily from ATP, which is not stored. Thus, cells need a continual supply of ATP to the extent that humans synthesize well over half their own weight in ATP daily. This is done by oxidizing glucose, free fatty acids (FFAs), amino acids (AAs), and ketone bodies. On average, the process of oxidizing fuels to form ATP is 40% efficient, with 60% lost as heat (Fig. 38-1). All fuels originally come from the diet–humans must eat to stay alive. Normally, people eat intermittently. Consequently, the use and distribution of fuels change over time.
Metabolic Phases
In general, there are four metabolic phases (Fig. 38-1): (1) the digestive or absorptive phase, which occurs during the 2 to 3 hours that it takes to digest a discrete meal; (2) the interdigestive or postabsorptive phase, which normally occurs between meals; (3) fasting, which most commonly occurs between the last snack before bedtime and breakfast (in fact, physicians refer to a blood value as “fasting,” e.g., “fasting blood glucose,” if the patient abstains from eating after midnight and has blood drawn about 8 AM; prolonged fasting and starvation are extreme forms of fasting); and (4) strenuous exercise or physical labor, which usually imposes an intense energy demand for a relatively short period (e.g., 1 hour).
A central feature of the utilization of different nutrients is the nature of cell-specific needs and capabilities. Cells with no or very few mitochondria cannot utilize AAs and FFAs for energy but must rely entirely on anaerobic glycolysis (see later). The brain, which continually accounts for about 20% of O2 consumption, cannot efficiently access circulating FFAs for energy. The brain converts most of its AA pool into neurotransmitters instead of oxidizing them for energy. This means that the brain and some other tissues are obligate glucose users. In other words, the function of the brain is critically dependent on circulating levels of blood glucose, much as it is on a continuous supply of O2. An acute fall in blood glucose levels below 50 mg/100 mL (i.e., hypoglycemia) leads to impaired central nervous system functions, including vision, cognition, and muscle coordination, as well as lethargy and weakness (Fig. 38-2). Severe hypoglycemia can ultimately lead to coma and death. Thus, a major role of the hormones involved in metabolic homeostasis is to maintain blood glucose levels above 60 mg/100 mL. Conversely, it is important that fasting blood glucose levels remain below 110 mg/100 mL. Indeed, the complications associated with poorly controlled diabetes mellitus have shown not only that too little blood glucose is incompatible with life but also that too much blood glucose imposes various stresses on cell function, increases morbidity, and shortens life (Fig. 38-2).
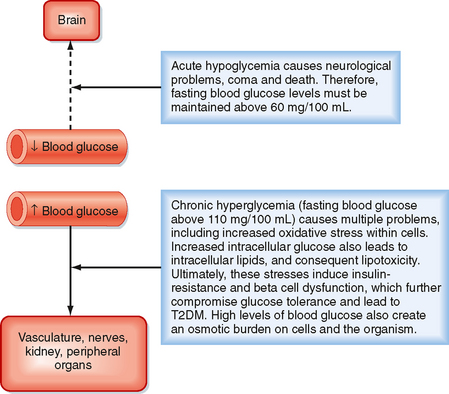
Figure 38-2 Importance of maintaining blood glucose within the normal range.
(Modified from Porterfield SP, White BA: Endocrine Physiology, 3rd ed. Philadelphia, Mosby, 2007.)
ATP SYNTHESIS
Making ATP from Carbohydrates
In the first phase (Fig. 38-3), glucose is transported across the cell membrane by bidirectional facilitative glucose transporters called GLUTs. Once inside the cell, glucose is prevented from exiting by phosphorylation to glucose-6-phosphate (G6P). This phosphorylation is catalyzed by hexokinases. The hexokinase that is expressed in the liver and pancreatic beta cells has low affinity for glucose (i.e., it transports glucose only when glucose is available at elevated concentrations) and is designated glucokinase.
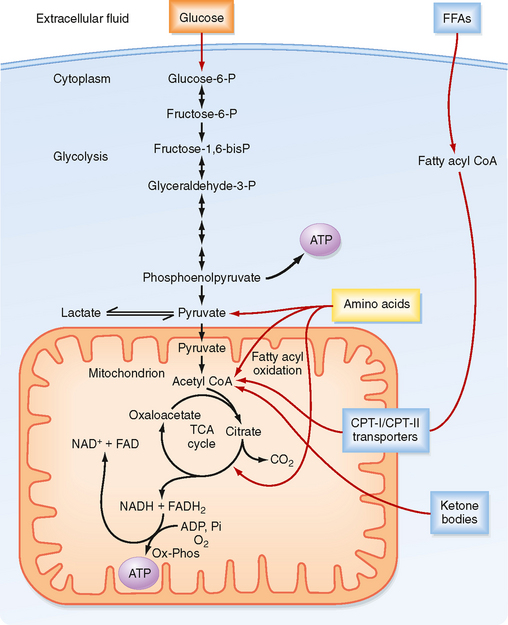
Figure 38-3 ATP is made from glucose, AAs, FFAs, and ketone bodies.
(Modified from Porterfield SP, White BA: Endocrine Physiology, 3rd ed. Philadelphia, Mosby, 2007.)
The second phase involves glycolysis (Fig. 38-3), which occurs in the cytoplasm. Glycolysis yields a net production of 2 mol of ATP/mol of glucose while consuming the required cofactor NAD+ by reducing it to NADH. In the presence of robust oxidative phosphorylation (relative to the rate of glycolysis), NADH is converted back to NAD+ in an O2-dependent manner, and pyruvate is the primary product of glycolysis (oxidative glycolysis). If the cell has no or very few mitochondria (e.g., erythrocytes, lens of the eye), oxidative phosphorylation cannot be carried out and used to oxidize NADH back to NAD+. In this case, the cell regenerates NAD+ by reducing pyruvate to lactate by the process of anaerobic glycolysis.
During the third process (Fig. 38-3), pyruvate enters the mitochondria and is converted to acetyl coenzyme A (acetyl CoA). Acetyl CoA is then further metabolized in the TCA cycle and the closely coupled process of oxidative phosphorylation via the electron transport chain. This second stage of oxidation yields almost 20 times more ATP than glycolysis does. Thus, the TCA cycle and oxidative phosphorylation are very efficient means of generating ATP from glucose. However, molecular O2 is required. This is why humans need to breathe air, and oxidative phosphorylation can proceed only as fast as the respiratory and cardiovascular systems can deliver O2 to tissues. Therefore, even tissues with mitochondria rely on anaerobic glycolysis for some needs. The process of oxidative phosphorylation is also a major contributor to the generation of reactive oxygen species (ROS), which impose oxidative stress that is harmful to cells.
Making ATP from Free Fatty Acids
The other two energy substrates, FFAs and AAs, bypass glycolysis and ultimately enter the TCA cycle/oxidative phosphorylation as pyruvate, acetyl CoA, or different components of the TCA cycle. FFAs are released from adipose tissue by lipolysis and circulate in blood bound to serum albumin. Transport proteins then translocate FFAs into cells. FFAs are metabolized in mitochondria by the repetitive, cyclic process of β oxidation (Fig. 38-3). This requires the transport of FFAs into the inner mitochondrial matrix by the carnitine palmitoyltransferase (CPT-I and CPT-II) system of transporters. Each cycle of β oxidation removes two carbon moieties at a time from FFA chains and generates a molecule of acetyl CoA, which is oxidized through the TCA cycle and oxidative phosphorylation. In addition to the generation of acetyl CoA, each cycle of β oxidation generates 1 molecule each of FADH2 and NADH, thereby producing up to 17 ATP molecules via oxidative phosphorylation. Thus, FFAs are a more efficient source of energy storage than carbohydrates in that the cell can obtain more ATPs per carbon from FFAs than from glucose.
Making ATP from Amino Acids
AAs can also be oxidized after transamination (transfer of their amino group to another molecule). The carbon skeletons of AAs converge on the TCA cycle by conversion to intermediates, including pyruvate, acetyl CoA, α-ketoglutarate, succinyl CoA, fumarate, and oxaloacetate (Fig. 38-3). The amino group of AAs can give rise to ammonia, a highly toxic substance. Thus, the use of AAs for energy must be coupled to the urea cycle in the liver, which converts ammonia to urea.
Making ATP from Ketone Bodies
Ketone bodies are four-carbon molecules that include acetoacetate and β-hydroxybutyrate. Ketone bodies do not exist in the diet at significant levels, as do carbohydrates, fats, and AAs. Rather, ketone bodies represent a fourth class of fuel that is synthesized from acetyl CoA in the liver and exported into the bloodstream for other organs to use. Extrahepatic tissues convert ketone bodies back to acetyl CoA by using succinyl CoA as a CoA donor and the enzyme thiophorase (Fig. 38-4). The liver itself lacks thiophorase and thus cannot use ketone bodies for its own energy needs.
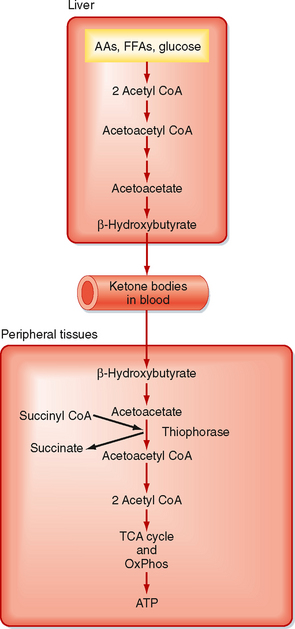
STORAGE FORMS OF ENERGY
Glycogen
In general, nutrients are stored during the fed state. Glucose can be stored as glycogen, which is a large polymer of glucose molecules. Once glucose is trapped in cells as G6P, it can be converted to glucose-1phosphate, which is then added to glycogen chains by two repetitive reactions. The primary, regulated enzyme in glycogenesis is glycogen synthase (Fig. 38-5).
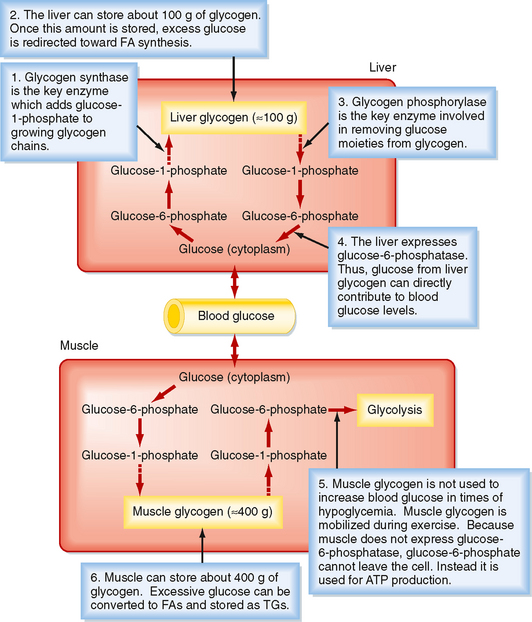
Figure 38-5 Glycogen synthesis and breakdown serve different needs in liver versus muscle.
(Modified from Porterfield SP, White BA: Endocrine Physiology, 3rd ed. Philadelphia, Mosby, 2007.)
During the interdigestive period, individual glucose moieties can be cleaved from glycogen and metabolized back to G6P (Fig. 38-5). The primary enzyme in glycogenolysis is called glycogen phosphorylase. In the liver, G6P can be further converted to glucose by glucose-6-phosphatase (G6Pase), and the glucose that is generated can be transported out of the cell by the bidirectional GLUT2 transporter. Thus, liver glycogen can directly contribute to blood glucose levels. Muscle does not express G6Pase, so glycogenolysis is linked to intramyocellular glycolysis. Muscle glycogen can contribute to blood glucose indirectly. Muscle glycolysis generates lactate, which is converted back to glucose by the liver through the process of gluconeogenesis (see later).
Triglyceride
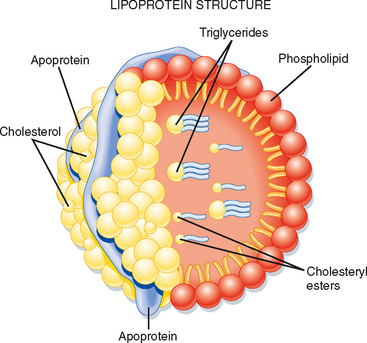
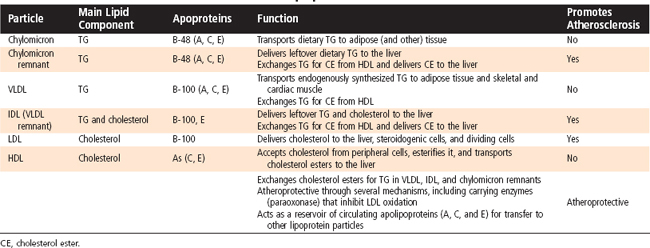
Triglyceride (TG) represents the storage form of nutrient lipid (e.g., FFAs). TG is obtained from the diet or synthesized endogenously by the liver in the face of caloric excess. Each molecule of TG is composed of three fatty acid chains in an ester linkage to each of the three carbons of glycerol. TG can be stored in most tissues, but only adipose tissue has evolved as a safe and efficient storage depot for TG. Significant TG accumulation in other organs (cardiac muscle, liver) can compromise their physiological functions and cause cell death. Accumulation of TG in skeletal muscle and liver also promotes insulin resistance and glucose intolerance. Thus, the body has developed transport mechanisms for the delivery of dietary and endogenously synthesized TGs to adipose tissue. These transport mechanisms involve the assembly of lipoprotein particles, which entails coating hydrophobic TG and cholesterol esters with relatively more hydrophilic (or amphipathic) free cholesterol and phospholipids (Fig. 38-6). Lipid-soluble vitamins (e.g., vitamins E, A, D, and K) also associate with lipoproteins. Specific apoproteins, as well as enzymes and transfer proteins, become associated with the surface of lipoprotein particles both before secretion and during transit in blood. The protein complement of lipoprotein particles is absolutely required for their specific function or functions and metabolic clearance. Lipoproteins are summarized in Table 38-2.
Dietary Triglyceride
Most of the TG stored in adipose tissue originates from the diet. Dietary TGs are digested by lipases in the intestinal lumen and are absorbed by intestinal cells as FFAs and 2-monoglycerides. These components are reassembled into TGs within enterocytes. The intestinal cells package TGs into a lipoprotein particle called a chylomicron, which enters the villar lymphatics (Fig. 38-7). The intestinal lymphatics bypass the hepatic portal circulation and the liver and empty into the general circulation. Once in blood, chylomicrons travel to adipose tissue, skeletal muscle, and cardiac muscle, where TGs are unloaded as FFAs and glycerol.
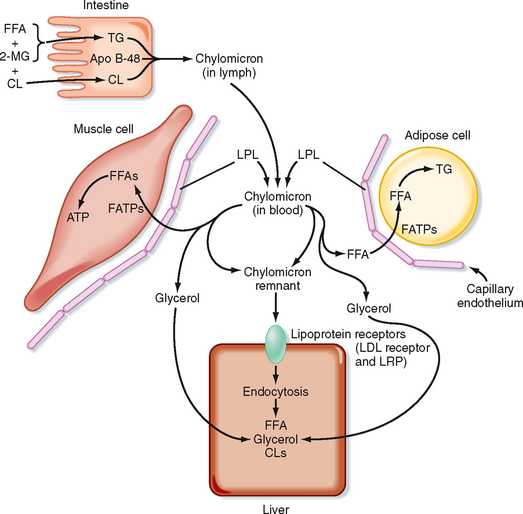
A primary apoprotein on chylomicrons is apo B-48. Secreted chylomicrons acquire additional apoproteins by transfer of proteins from high-density lipoproteins (HDLs) in blood. For example, Apo C-II is an apoprotein that is exchanged between HDL and chylomicrons. Apo C-II acts as an activator/cofactor of the enzyme lipoprotein lipase (LPL), which digests circulating chylomicrons. LPL is synthesized by adipocytes and muscle cells. It is secreted and ultimately translocated to the apical surface of the endothelium lining neighboring capillaries, to which LPL remains noncovalently attached by heparin sulfate proteoglycans. Dozens of LPL molecules attach to and digest lipoprotein particles, thereby releasing FFAs and glycerol (Fig. 38-7). Several fatty acid transport proteins are involved in the transport of FFAs from the apical surface of endothelial cells to the cytoplasm of neighboring cells. Once FFAs enter a cell, they are immediately converted to fatty acyl CoAs. In skeletal and cardiac muscle, fatty acyl CoAs are oxidized for production of ATP. In adipocytes, FFAs are stored in the form of TG. Esterification of the first fatty acyl chain requires glycerol-3-phosphate (G3P). Adipocytes do not express glycerol kinase and consequently cannot synthesize G3P directly from the glycerol released from chylomicrons. Instead, adipocytes generate G3P from intermediates of glycolysis. Partially digested, TG-depleted chylomicrons are called chylomicron remnants. These are cleared by the liver through the process of receptor-mediated endocytosis, which requires another apoprotein, apo E. Multiple apo E proteins are transferred to a chylomicron from HDL and bind to the low-density lipoprotein (LDL) receptor and LDL receptor–related protein (LRP) on hepatocyte membranes.
Endogenously Synthesized Triglyceride
TGs can also be synthesized from glucose and other precursors of acetyl CoA (Fig. 38-8). This occurs during high caloric intake when liver and muscle glycogen stores are saturated and the supply of glucose exceeds the need for synthesis of ATP (e.g., during the development of diet-induced obesity). The primary site of endogenous FFA and TG synthesis in humans is the liver, usually in response to high levels of glucose. Glucose is metabolized to acetyl CoA and then to citrate in the first reaction of the TCA cycle. However, the presence of high ATP and NADH levels in the well-fed state inhibits progression of the TCA cycle and causes intramitochondrial levels of citrate to accumulate. Citrate is then translocated to the cytoplasm, where it is converted back to cytosolic acetyl CoA and oxaloacetate. Once in the cytoplasm, acetyl CoA can enter fatty acyl CoA and TG synthesis (see later). Fatty acyl CoAs are esterified to G3P to form monoglycerides, diglycerides, and finally TGs. TGs are not normally stored in the liver to a large extent but are transferred to adipose tissue. Thus, TGs must be packaged by the liver into lipoprotein particles called very-low-density lipoproteins (VLDLs) before being secreted into blood. Like chylomicrons, VLDLs contain a core of very hydrophobic TG and cholesterol esters and a covering of amphipathic phospholipids and free cholesterol. The VLDL particle also contains apo B-100. After secretion, VLDLs acquire other proteins from circulating HDL particles, including apo C-II and apo E, and are digested by LPL within the capillary beds of adipose tissue, as well as skeletal and cardiac muscle (Fig. 38-8).
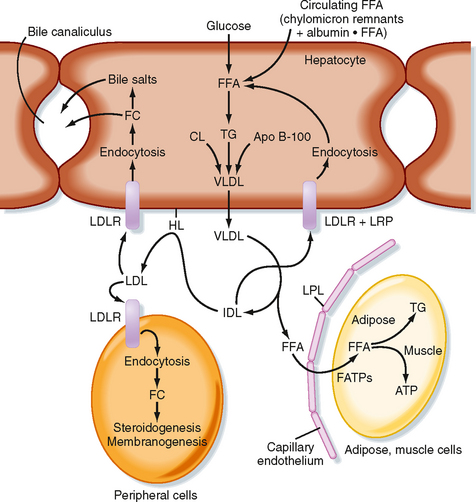
Partially LPL-digested VLDL particles are called VLDL remnants, or intermediate-density lipoprotein (IDL) particles (Fig. 38-8). IDL has two fates. First, IDL is removed from the circulation by receptor-mediated endocytosis by the LDL receptor (via binding to apo B-100 and apo E) and LRP (via binding to apo E) at the liver. Efficient endocytosis of IDL is dependent on multiple copies of apo E being associated with the remnant particle. Second, IDL is further digested by the ectoenzyme hepatic lipase. This delivers FFAs and glycerol to the liver and transforms the IDL into a TG-poor, cholesterol-rich LDL particle.
Low-Density Lipoprotein and Cholesterol Economy
LDL particles deliver cholesterol to cells through binding of apo B-100 to the LDL receptor, followed by receptor-mediated endocytosis. In the transition of IDL to LDL, the LDL loses apo E. This means that LDL cannot be cleared from blood through apo E–dependent binding to LRP, only through apo B-100–dependent binding to the LDL receptor. Quantitatively, the primary site of LDL endocytosis is the liver, which is also the site of cholesterol excretion. About 1 g of cholesterol is excreted daily by the liver–50% as cholesterol and 50% as bile salts. The liver is also the primary site of cholesterol synthesis. Importantly, the synthesis of cholesterol and uptake of LDL cholesterol are highly regulated in a negative-feedback loop. Therefore, the daily amount of cholesterol synthesis (about 1 g) is modulated by the amount absorbed from the diet (about 250 mg/day), so changes in dietary cholesterol normally have a relatively small effect on total circulating and LDL cholesterol.
High-Density Lipoprotein and Reverse Cholesterol Transport
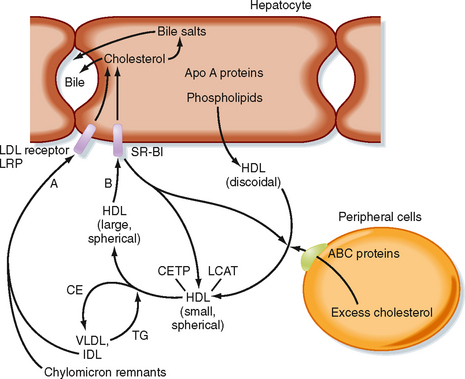
Because cells cannot break cholesterol down, they intermittently need to discharge cholesterol (Fig. 38-9). There is also a need for macrophages that ingest oxidized LDL to rid themselves of excess cholesterol before they become foam cells and die. The efflux of cholesterol from cells is facilitated by ATP-binding cassette (ABC) proteins, most notably ABCA1. The cholesterol that is transferred out of cells is accepted by nascent HDL. Nascent HDL is produced by the liver and small intestine and is composed of apo A-I, phospholipids (primarily lecithin), and the enzyme lecithin-cholesterol acyltransferase (LCAT). LCAT esterifies cholesterol, and the cholesterol esters accumulate within the center of the maturing spherical HDL (HDL3). Mature HDL can return cholesterol to the liver for excretion (i.e., reverse cholesterol transport) via two pathways. First, HDL can transfer cholesterol esters to TG-rich VLDL, IDL, and chylomicron remnants through the action of an HDL-associated protein, cholesterol ester transfer protein (CETP). This is done in exchange for TG, which produces a larger HDL particle (HDL2). The cholesterol-enriched IDL and chylomicron remnants are then endocytosed by the liver through apo E–dependent binding to the LDL receptor and LRP (A in Fig. 38-9). The second pathway involves apo A-I–dependent binding to the scavenger receptor BI (SR-BI) on the hepatocyte membrane (B in Fig. 38-9). This allows the transfer of cholesterol esters from HDL into the hepatocyte membrane. Cholesterol esters are then cleaved by hepatic hormone-sensitive lipase, and the free cholesterol enters the bile salt pathway or is excreted as cholesterol. Larger, TG-enriched HDL is first processed by hepatic lipase, which decreases its size and enhances its binding to SR-BI.
Catabolism of Triglycerides in Adipose Cells
During a fast, TGs are catabolized back to FFAs and glycerol. This is initiated by the action of a hormone-sensitive lipase, followed by additional lipases that remove the second and third fatty acyl groups. The net amount of TG versus FFAs in adipose tissue is thus determined by the balance of TG synthesis and lipolysis, which is extremely sensitive to hormonal signals. Hydrophobic FFAs are transported in blood mostly in FFA-albumin complexes. FFAs are actively transported into cells, which divert FFAs into β oxidative pathways for energy or, in the case of the liver, into ketone bodies (Fig. 38-4). This latter fate of FFAs is important during a prolonged fast because unlike FFAs, ketone bodies in sufficient levels can cross the blood-brain barrier.
< div class='tao-gold-member'>
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


