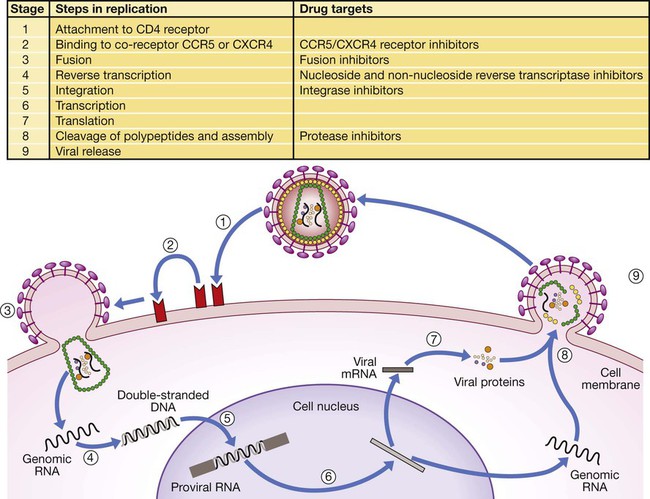
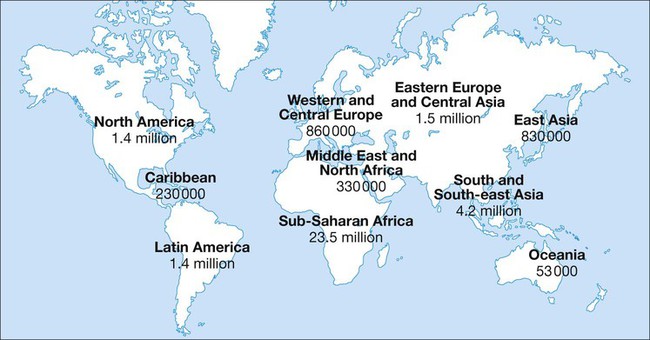
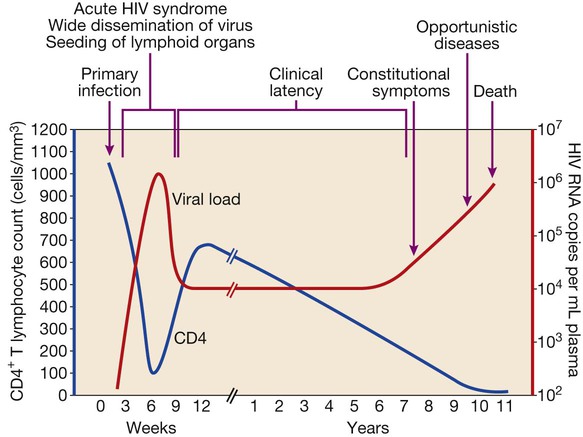
*These conditions are in WHO stage 4 but not in CDC category C. In 2011 it was estimated that there were 34.2 million people living with HIV/AIDS, 2.5 million new infections and 1.7 million deaths (Fig. 14.1). Globally, new infections have declined by 20% over the last 10 years. Not all regions have experienced reductions in new infections and the dominant modes of transmission also vary regionally (Box 14.1). Expanding access to combination antiretroviral therapy (ART) has resulted in a 24% decline in global AIDS-related deaths since the peak in 2005. The improved life expectancy on ART has resulted in an increase in the number of people living with HIV. Despite these encouraging epidemiological data, HIV remains an important cause of death globally and has caused over 30 million deaths since the epidemic started. HIV has had a devastating effect in sub-Saharan Africa, particularly in southern African where average life expectancy of the general population fell to below 40 years. (IDU = intravenous drug users; MSM = men who have sex with men) HIV is transmitted by sexual contact, by exposure to blood (e.g. injection drug use, occupational exposure in health-care workers) and blood products, or to infants of HIV-infected mothers (who may be infected in utero, perinatally or via breastfeeding). Worldwide, the major route of transmission is heterosexual. The risk of contracting HIV after exposure to infected body fluid is dependent on the integrity of the exposed site, the type and volume of fluid, and the level of viraemia in the source person. The approximate transmission risk after exposure is given in Box 14.2. Factors that increase the risk of transmission are listed in Box 14.3. HIV can only infect cells bearing the CD4 receptor; these are T-helper lymphocytes, monocyte–macrophages, dendritic cells, and microglial cells in the central nervous system (CNS). Entry into the cell commences with binding of gp120 to the CD4 receptor (stage 1, Fig. 14.2), which results in a conformational change in gp120 that permits binding to one of two chemokine co-receptors (CXCR4 or CCR5: stage 2). The chemokine co-receptor CCR5 is utilised during initial infection, but later on the virus may adapt to use CXCR4. Individuals who are homozygous for the CCR5 delta 32 mutation do not express CCR5 on CD4 cells and are immune to HIV infection. Chemokine receptor binding is followed by membrane fusion and cellular entry involving gp41 (stage 3). After penetrating the cell and uncoating, a deoxyribonucleic acid (DNA) copy is transcribed from the RNA genome by the reverse transcriptase (RT) enzyme (stage 4) that is carried by the infecting virion. Reverse transcription is an error-prone process and multiple mutations arise with ongoing replication, which results in considerable viral genetic heterogeneity. Viral DNA is transported into the nucleus and integrated within the host cell genome by the integrase enzyme (stage 5). Integrated virus is known as proviral DNA and persists for the life of the cell. Cells infected with proviral HIV DNA produce new virions only if they undergo cellular activation, resulting in the transcription of viral messenger RNA (mRNA) copies (stage 6), which are then translated into viral peptide chains (stage 7). The precursor polyproteins are then cleaved by the viral protease enzyme to form new viral structural proteins and enzymes (stage 8). These then migrate to the cell surface and are assembled using the host cellular apparatus to produce infectious viral particles, which bud from the cell surface, incorporating the host cell membrane into the viral envelope (stage 9). The mature virion then infects other CD4 cells and the process is repeated. CD4 lymphocytes that are replicating HIV have a very short survival time of about 1 day. It has been estimated that in asymptomatic HIV-infected people, more than 1010 virions are produced and 109 CD4 lymphocytes destroyed each day. Globally, the trend is towards universal HIV testing, rather than testing patients at high risk or those with manifestations of HIV infection only. However, in the UK, testing is still targeted (Box 14.4). HIV is diagnosed by detecting host antibodies either by using rapid point-of-care tests or in the laboratory, where enzyme-linked immunosorbent assay (ELISA) tests are usually done. Most tests detect antibodies to both HIV-1 and HIV-2. A positive antibody test from two different immunoassays is sufficient to confirm infection. Western blot assays can also be used to confirm infection, but they are expensive and sometimes yield indeterminate results. Screening tests often include a test for p24 antigen in addition to antibodies, in order to detect patients with primary infection before the antibody response occurs. Nucleic acid amplification tests (usually PCR) to detect HIV-RNA are used to diagnose infections in infants of HIV-infected mothers, who carry maternal antibodies to HIV for up to 15 months irrespective of whether they are infected, and to diagnose primary infection before antibodies have developed. (PCR is more sensitive than p24 antigen detection, but p24 is more widely available.) • All those presenting with a possible primary HIV infection or where HIV enters the differential diagnosis • All sexual partners of an HIV-positive individual • All MSM and female sexual contacts of MSM • All with a history of injecting drug use • All from a country of high HIV prevalence • All who report sexual contact abroad or in the UK with individuals from a country of high HIV prevalence (> 1%) The purpose of HIV testing is not simply to identify infected individuals, but also to educate people about prevention and transmission of the virus. Counselling is essential both before HIV testing and after the result is obtained (Boxes 14.5 and 14.6). There are major advantages to using rapid point-of-care HIV tests in that pre- and post-test counselling can be done at the same visit. Counselling should always be given in the client’s home language. A number of baseline investigations should be done at the initial medical evaluation (Box 14.7). The extent of these investigations will depend on the resources available. The level of viraemia is measured by quantitative PCR of HIV-RNA, known as the viral load. Determining the viral load is important for monitoring responses to ART (p. 407) and also has some prognostic value before starting ART. However, many low-income countries are unable to afford viral load measurements. People with high viral loads (e.g. > 100 000 copies/mL) experience more rapid declines in CD4 count, while those with low viral loads (< 1000 copies/mL) usually have slow or even no decline in CD4 counts. There is little point in repeated measurements of viral load before starting ART, as viral loads remain at a relatively stable plateau after primary infection (Fig. 14.3). Clinical staging of patients should be done at the initial medical examination, as it provides prognostic information and is a key criterion for initiating ART and prophylaxis against opportunistic infections. Two clinical staging systems are used internationally (p. 389). In both systems, patients are staged according to the most severe manifestation and do not improve their classification. For example, a patient who is asymptomatic following a major opportunistic disease (AIDS) remains at stage 4 or category C of the WHO and CDC systems respectively, and never reverts to earlier stages. Finally, patients do not always progress steadily through all stages and may present with AIDS, having previously been asymptomatic. Primary infection is symptomatic in more than 50% of cases, but the diagnosis is often missed. The incubation period is usually 2–4 weeks after exposure. The duration of symptoms is variable, but is seldom longer than 2 weeks. The clinical manifestations (Box 14.8) resemble a glandular fever-type illness, but the presence of maculo-papular rash or mucosal ulceration strongly suggests primary HIV infection rather than the other viral causes of glandular fever (p. 320). In infectious mononucleosis due to other viruses, rashes generally only occur if aminopenicillins are given. Atypical lymphocytosis occurs less frequently than in Epstein–Barr virus (EBV) infection. Transient lymphopenia, including CD4 lymphocytes, is found in most cases (see Fig. 14.3), which may result in opportunistic infections, notably oropharyngeal candidiasis. Major opportunistic infections like Pneumocystis jirovecii pneumonia (PJP) may rarely occur. Thrombocytopenia and moderate elevation of liver enzymes are commonly present. The differential diagnosis of primary HIV includes acute EBV, primary cytomegalovirus (CMV) infection, rubella, primary toxoplasmosis and secondary syphilis. Viraemia peaks during primary infection and then drops as the immune response develops, to reach a plateau about 3 months later. The level of viraemia post-seroconversion is a predictor of the rate of decline in CD4 counts, which is highly variable and explained in part by genetic factors affecting the immune response. The median time from infection to the development of AIDS in adults is about 9 years (see Fig. 14.3). A small proportion of untreated HIV-infected people are long-term non-progressors with CD4 counts in the reference range for 10 years or more. Some long-term non-progressors have undetectable viral loads and are known as ‘elite controllers’. AIDS is defined by the development of specified opportunistic infections, cancers and severe manifestations of HIV itself (p. 389). CDC category C is the most widely used definition of AIDS. WHO updated its classification more recently and added a few conditions of similar prognosis to its stage 4 disease. Box 14.9 outlines the correlation between CD4 count and HIV-related diseases.
HIV infection and AIDS
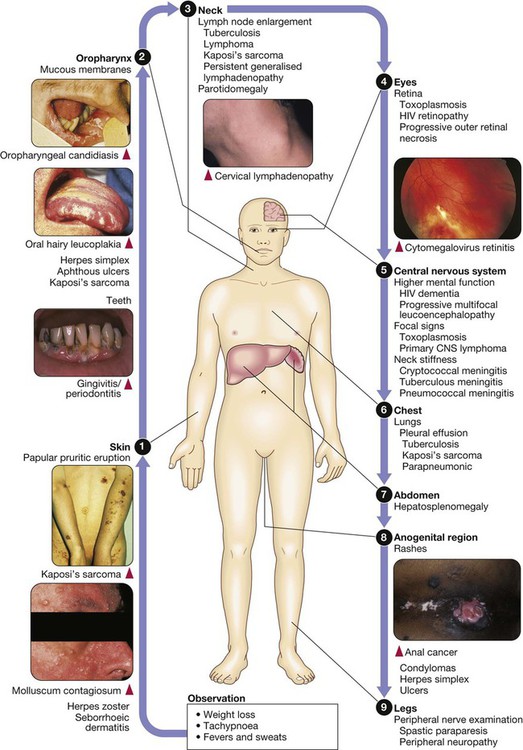
Clinical examination in HIV disease
 HIV clinical staging classifications
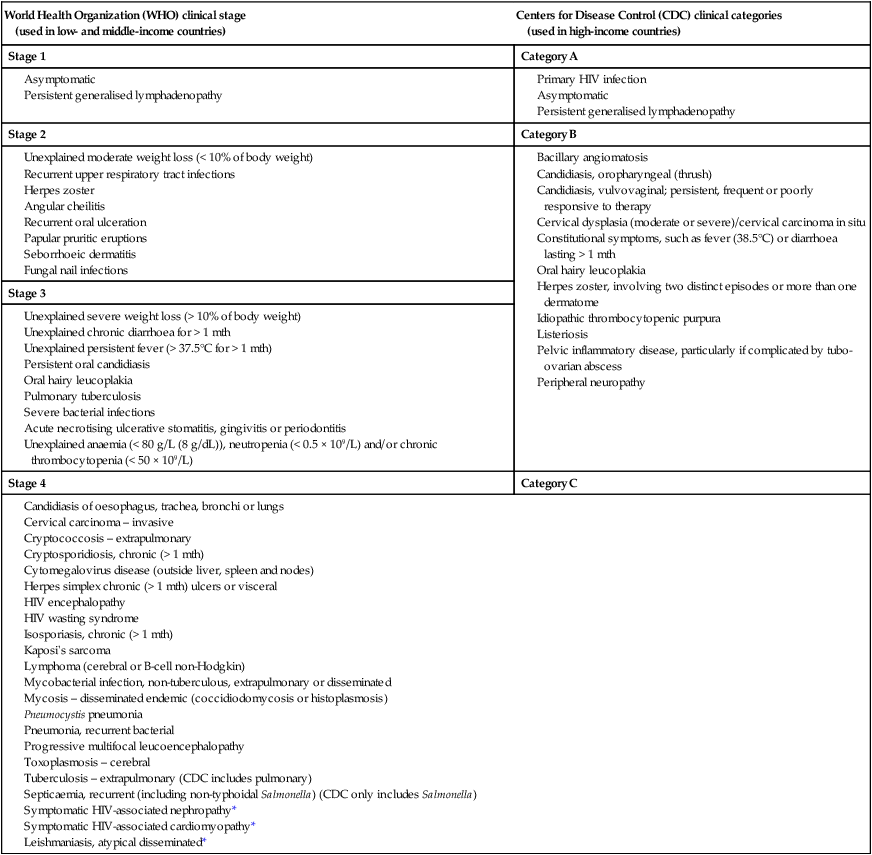
HIV clinical staging classifications
Epidemiology
Global epidemic and regional patterns
 14.1 Regional HIV incidence and transmission, 2001–2011
14.1 Regional HIV incidence and transmission, 2001–2011
Region
HIV incidence
Dominant transmission
Sub-Saharan Africa
Decreasing
Heterosexual
South, South-east and East Asia
Decreasing
IDU, heterosexual
Oceania
Decreasing
Heterosexual
Caribbean
Decreasing
Heterosexual
Latin America
Stable
MSM
North America
Stable
MSM
Western and Central Europe
Stable
MSM
Eastern Europe and Central Asia
Increasing
IDU
Middle East and North Africa
Increasing
IDU, MSM
Modes of transmission
Virology and immunology
Diagnosis and investigations
Diagnosing HIV infection
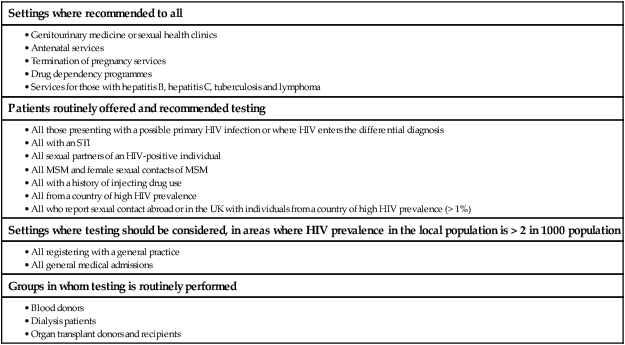
 14.4 HIV testing in the UK
14.4 HIV testing in the UK
Settings where recommended to all
Patients routinely offered and recommended testing
Settings where testing should be considered, in areas where HIV prevalence in the local population is > 2 in 1000 population
Groups in whom testing is routinely performed
 14.5 How to carry out pre-test counselling
14.5 How to carry out pre-test counselling
 14.6 How to carry out post-test counselling
14.6 How to carry out post-test counselling
Viral load and CD4 counts
Viral load
Natural history and staging of HIV
Primary infection
Asymptomatic infection
Acquired immunodeficiency syndrome
 14.2
14.2 14.3
14.3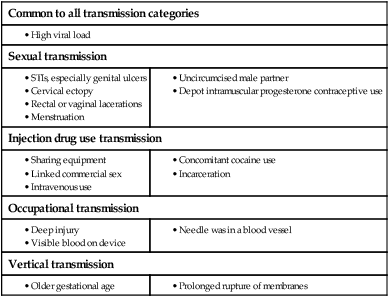
 14.7
14.7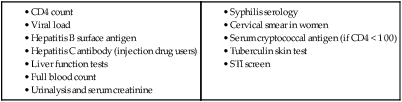
 14.8
14.8
 14.9
14.9


