HIV-1: Pathogenesis, Clinical Manifestations, and Treatment
Daniel R. Kuritzkes
Richard A. Koup
Human immunodeficiency virus (HIV) is essentially an infection of the immune system. The main clinical manifestation of infection is the progressive and ultimately profound defect in cell-mediated immune responses that are essential for protection against a variety of normally innocuous agents that become the major sources of morbidity and mortality. Over the 30 years since the original identification of a new syndrome that was ultimately shown to be caused by HIV,55 the infection has become a grave humanitarian crisis with far ranging impact on international political and economic stability, and contributes greatly to global health inequalities in terms of access to life-saving therapies.
Epidemiology
The initial clues to the emerging HIV epidemic came from two linked observations in 1980–1981. One was from front-line physicians, predominantly in New York and California, who were confronted with previously healthy young men presenting with symptoms of profound unexplained immune deficiency. The other was at the Centers for Disease Control and Prevention (CDC) in Atlanta, which noted a marked increase in the number of requests for pentamidine, a drug used to treat an infection associated with severe immunodeficiency. The link between these observations was the sudden upsurge in cases of Pneumocystis jirovecii (formerly P. carinii) pneumonia being diagnosed in young gay men, an opportunistic infection previously seen in the setting of immune suppression accompanying cancer chemotherapy, but now, for the first time, appearing in persons with no obvious disposition to develop immune deficiency.
By 1984, the viral etiology of what became known as HIV type 1 (HIV-1) infection had been confirmed through the isolation of a lymphotropic retrovirus from infected persons, and the detection of antibodies in persons at risk.22,127,322 It was clear that certain groups were particularly at risk, including gay men, injection drug users, recipients of blood transfusions, and hemophiliacs, and that the mechanisms of transmission began to be defined.81,189 By April 1985, a blood test for detection of antibodies was licensed, and the extent of the epidemic began to unfold. Since the initial detection of the epidemic in the United States and Europe, it has grown to involve an estimated 60 million persons worldwide, with 5 million new infections per year, with epidemics expanding in eastern Europe, south Asia, and China317 (Fig. 50.1). In Southern Africa, less than 1% of the population was infected in 1990, when large scale surveillance programs were initiated, whereas by 2004 antenatal screening surveys detected a prevalence well in excess of 30% in some areas.308
Origin of HIV
The global epidemic of HIV-1 is the result of a cross-species infection of humans by a chimpanzee lentivirus, simian
immunodeficiency virus (SIVcpz), which occurred in west central Africa.131 Chimpanzees, in turn, appear to have acquired SIVcpz sometime after their divergence into multiple subspecies. Pan troglodytes and P.t. schweinfurthii are naturally infected, whereas other closely related species are not, suggesting that SIVcpz is unlikely to have coevolved with its natural host.366 How the virus initially arose as a new virus in chimpanzees remains unclear, but it likely occurred following transmission of two other ancestral SIV species from monkeys,366 perhaps because chimpanzees hunt monkeys for food. Of note, SIVcpz is not an asymptomatic infection in chimpanzees; the epidemiologic evidence documents decreased survival in chimpanzee troops infected by SIVcpz.209 In contrast, other SIV infections appear to be largely asymptomatic in their natural monkey hosts (i.e., SIVsmm in sooty mangabeys) and do not lead to immune deficiency or decreased life span. Experimental transmission of these viruses to susceptible non-natural hosts can result in progressive and profound immunodeficiency and acquired immunodeficiency disease (AIDS).354 In fact, the transmission of SIVsmm in West Africa is the origin of the HIV-2 epidemic in humans.354 Although the reasons for differences in transmissibility and pathogenicity during cross-species transmission are not fully understood, it is clear that innate host defenses such as those imparted by apolipoprotein B messenger RNA (mRNA)–editing enzyme, catalytic polypeptide-like 3G (APOBEC3G), and tripartite motif 5 alpha (TRIM-5α) play a role and create barriers that viruses must overcome in order to become established and survive within a new host species (see Chapter 49).
immunodeficiency virus (SIVcpz), which occurred in west central Africa.131 Chimpanzees, in turn, appear to have acquired SIVcpz sometime after their divergence into multiple subspecies. Pan troglodytes and P.t. schweinfurthii are naturally infected, whereas other closely related species are not, suggesting that SIVcpz is unlikely to have coevolved with its natural host.366 How the virus initially arose as a new virus in chimpanzees remains unclear, but it likely occurred following transmission of two other ancestral SIV species from monkeys,366 perhaps because chimpanzees hunt monkeys for food. Of note, SIVcpz is not an asymptomatic infection in chimpanzees; the epidemiologic evidence documents decreased survival in chimpanzee troops infected by SIVcpz.209 In contrast, other SIV infections appear to be largely asymptomatic in their natural monkey hosts (i.e., SIVsmm in sooty mangabeys) and do not lead to immune deficiency or decreased life span. Experimental transmission of these viruses to susceptible non-natural hosts can result in progressive and profound immunodeficiency and acquired immunodeficiency disease (AIDS).354 In fact, the transmission of SIVsmm in West Africa is the origin of the HIV-2 epidemic in humans.354 Although the reasons for differences in transmissibility and pathogenicity during cross-species transmission are not fully understood, it is clear that innate host defenses such as those imparted by apolipoprotein B messenger RNA (mRNA)–editing enzyme, catalytic polypeptide-like 3G (APOBEC3G), and tripartite motif 5 alpha (TRIM-5α) play a role and create barriers that viruses must overcome in order to become established and survive within a new host species (see Chapter 49).
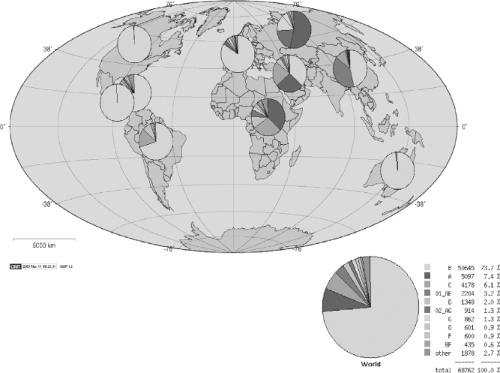 Figure 50.1. Global subtype distribution of human immunodeficiency virus type 1 (HIV-1) clades as reported in the Los Alamos Database. |
At least three distinct cross-species transmissions from chimpanzees to humans are thought to have occurred, giving rise to three highly divergent genetic lineages of infection, termed groups M, N, and O.158,302,372 Group M is by far the most widespread, accounting for most cases globally. Because the greatest genetic diversity in group M is found in Kinshasa, Zaire, the epidemic likely began in this region of Africa. The approximate date of introduction of the M group into the human population is estimated to be around 1931, based on supercomputer analysis of large numbers of sequenced HIV isolates, assuming a constant rate of evolution.218 This approach has been validated by successfully timing the earliest historic isolate, which was sequenced from a stored plasma sample obtained in 1959 from a person who died of AIDS in Manchester, England.77
Current Global Distribution
The most heavily affected continent at the present time is Africa, with an estimated 25 million persons currently infected with HIV, and at least 3 million with advanced disease in need of treatment. HIV infections have increased markedly over the last decade, despite knowledge of the mechanisms of transmission. Expanding epidemics in Eastern Europe, South Asia, and China may ultimately eclipse the African epidemic in terms of sheer numbers of infected persons. The viruses fueling these epidemics vary according to geographic region, with clade C
virus being the most prevalent worldwide and clade B being currently the most prevalent in the United States and Europe. Clades differ one from another by up to 35%; within a single clade variation between isolates can be as great as 20%. Mistakes in reverse transcription, which occur because the viral reverse transcriptase lacks an editing function, leads to variability of individual virus sequences within a single infected person that can exceed what is seen with other viruses (e.g., influenza) over the course of a global epidemic.135
virus being the most prevalent worldwide and clade B being currently the most prevalent in the United States and Europe. Clades differ one from another by up to 35%; within a single clade variation between isolates can be as great as 20%. Mistakes in reverse transcription, which occur because the viral reverse transcriptase lacks an editing function, leads to variability of individual virus sequences within a single infected person that can exceed what is seen with other viruses (e.g., influenza) over the course of a global epidemic.135
Table 50.1 Risk of Transmission of HIV-1 | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Mechanisms of Transmission
Most cases of HIV infection worldwide are the result of sexual transmission, but important modes of transmission also include parenteral transmission and transmission from mother to infant. The actual risk of transmission per exposure varies widely (Table 50.1). For sexual transmission, the risk of male to male transmission is greater than the risk of heterosexual transmission, and is highest in persons practicing receptive anal intercourse.348 Overall, the greatest risk of transmission is from contaminated blood transfusions, with 95 of 100 persons becoming infected, although transfusion-related infections are rare now that blood is routinely screened for this pathogen.361 The rate of transmission depends on the inoculum size delivered, but perhaps also on local mucosal defense mechanisms that modulate infection but remain to be defined. Population studies performed in Africa suggest that there is a threshold viral load (∼1,500 RNA copies/mL plasma) below which the likelihood of sexual transmission is greatly reduced.150,325
Mathematical modeling suggests that the published risks of sexual transmission, which come from longitudinal studies of persons in identified risk groups, are too low to account for the huge numbers of infections worldwide and for the rapid increase in infections that occur after puberty. In South Africa, for example, 4% of 16-year-old girls are HIV seropositive, whereas by age 21 this number increases to 31%.308 Coital frequency estimates in these same age groups fail to account for this rapid increase in infections. More recent data suggest that transmission is amplified during periods of increased genital tract viral shedding.128 Increased shedding certainly occurs at the time of acute infection, which has been shown through a study of more than 15,000 persons in 56 villages in Uganda to be a particularly important period for transmission and, thus, has important public health implications.411 An important aspect of this study was that it followed couples who were both seronegative at the time of enrollment, so it was not potentially selecting for discordant couples who might have already demonstrated some intrinsic resistance to transmission. Other factors that may amplify the risk of transmission include other co-infections (e.g., malaria and tuberculosis, sexually transmitted diseases [STDs], or in particular co-transmission of STD and HIV).71 Estimated transmission probability likely also depends on genetic factors, such as density of the HIV co-receptor CCR5 on cervical cells.58 In addition to using epidemiologic data, much has been learned about transmission across a mucosal barrier by studying the SIV system, where it has been shown that CD4 T cells are the first cells to be infected (as opposed to antigen-presenting cells), and that there is a small temporal window of opportunity in which antiviral therapy or immune responses can block viral dissemination.159,433
Pathogenesis
Human immunodeficiency virus directly infects and kills cells that are critical for effective immune responses. Through direct interaction between the viral envelope and its cellular receptor, CD4 and the chemokine coreceptor, CCR5 or CXCR4 (see Chapter 49), this virus infects key cells of the adaptive immune response, explaining the clinical manifestations of disease being profound immune suppression. The course of disease varies enormously among infected persons. The time from acute infection to the development of AIDS, defined by a CD4 cell count of less than 200 cells/μL or the appearance of AIDS-defining opportunistic infections or cancers, can be as rapid as 6 months.310 Other persons have been known to be infected for more than 25 years and to maintain normal CD4 cell levels and exhibit no evidence of CD4-cell decline or immune deficiency, despite never having been treated with anti-HIV medications.271 A precise explanation for these differences in disease development remains elusive, but increasing evidence suggests that early events at the time of acute infection, together with viral and host genetics, play a determining role in the clinical outcome of disease.
Acute Infection and CD4 Cell Depletion
Ever since the analysis of viral load in the Multicenter AIDS Cohort Study (MACS) dataset in 1996,267 it has been clear that the course of HIV infection can be predicted within the first 6 to 12 months based on plasma viral load, and that CD4 count at this time also independently helps to determine whose disease will progress rapidly and whose will not.246 The median viral load in plasma at the time of peak viremia during acute infection is approximately 106 to 107 RNA copies/mL, which drops to a mean set point of 30,000 copies/mL within the first 6 to 12 months of infection. Longitudinal studies from the MACS, which were initiated before the availability of effective antiviral therapy, revealed the interquartile ranges of viral load and the relationship to risk of disease progression (Fig. 50.2). Interestingly, at 1 year after infection, only a fivefold difference
was seen in plasma viremia between the quartile that does the best and the quartile that does the worst.246
was seen in plasma viremia between the quartile that does the best and the quartile that does the worst.246
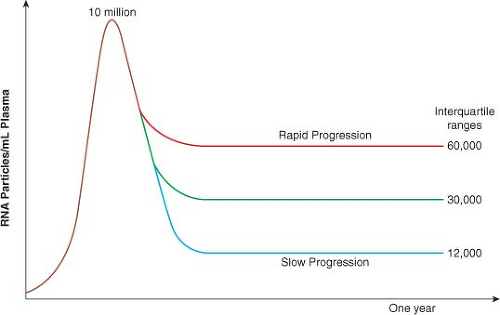 Figure 50.2. Interquartile ranges of viral load within one year of acute human immunodeficiency virus (HIV) infection. (Adapted from Lyles RH, Munoz A, Yamashita TE, et al. Natural history of human immunodeficiency virus type 1 viremia after seroconversion and proximal to AIDS in a large cohort of homosexual men. J Infect Dis 2000;181:872–880.) |
Studies in both humans infected with HIV43,264 and the SIV macaque model234,261 provide important new insights into why the period of acute infection plays such a defining role in ultimate disease progression, and why the immune system ultimately fails to control HIV in most infected persons. Infection and depletion of large numbers of both resting and activated memory CD4 cells in the gut during primary infection may be what fuels the initial peak viremia, and likely places constraints on subsequent adaptive responses43,264 (Fig. 50.3). Details of what is happening at a cellular level in acute infection have come from experimental infection of macaques with SIV. At the time of acute SIV infection, massive infection occurs of gut-associated lymphoid tissue, with between 30% and 60% of all gut-associated CD4 cells becoming productively infected, leading to massive depletion of these cells in only 4 days.261 The memory cell population, including both activated and resting memory cells, seems most vulnerable.234 In the earliest stages of infection, more than half of all memory CD4+ T cells are lost, and the body is left to fight the prolonged chronic phase of infection with a severely constricted repertoire of CD4 T helper cells. These experiments in the SIV model are almost certainly representative of what is happening in acute HIV infection in humans, where the initial plasma viral load averages 1 to 10 million RNA copies/mL. Whether the subsequent decline to a steady state results from active inhibition by innate and adaptive immune responses to HIV, or a decrease in the number of cells available to support viral replication, remains an area of dispute.312
Most of the CD4 T cells that are depleted from the gut during acute HIV or SIV infection are T helper cells that produce interleukin 17 (IL-17) (Th17 cells). These cells are crucial for maintaining the integrity of the mucosal barrier, and their depletion by HIV or SIV leads to loss of epithelial integrity within the gut mucosa, thereby allowing translocation of gut-associated microbial products into the systemic circulation.110 These products are a major driver of the systemic immune activation that is a predominant feature of all pathogenic HIV and SIV infections.41,42 The loss of CD4 T cells from the gut during acute infection therefore has two effects upon the immune system: it depletes a major portion of the immune reserve in the form of memory CD4 T cells, and it allows microbial translocation, which establishes a state of chronic immune activation that further promotes systemic HIV infection and replication.
Establishment of a Latent Viral Reservoir
During the period of acute infection, a stable reservoir of HIV-infected resting memory CD4 cells is established that harbors replication-competent, integrated provirus.63,116 This occurs whether or not persons are started on treatment in the initial symptomatic stage of infection. Because proviruses that form this latent reservoir are not transcriptionally active, no viral proteins or enzymes are produced. These infected cells therefore are protected from the effects of antiviral drug therapy and from immune attack. This latent reservoir is present whether the viral load is high or low, and it persists even after prolonged highly active antiviral therapy.115 A rough estimate is that about 1 in 1 × 106 CD4 cells harbors integrated latent provirus.434 The half-life of infected cells is such that it would take more than 70 years to achieve viral eradication by gradual senescence of these cells even with no blips in viremia on therapy,434 a situation that is yet to be achieved (see below).
Host Immunity
Recovery from many human viral infections is not associated with the eradication of infection, but rather results from immune-mediated containment. For example, persons who develop herpes zoster caused by varicella zoster virus infection ultimately contain the initial infection with a potent immune response.92 The virus remains present for the rest of the life of the infected individual; in most persons, however, it is controlled by an effective immune response. Although HIV infection is associated with progressive and ultimately profound immune suppression, a highly variable course of disease is seen among infected persons. After more than 25 years since the humoral and cellular
immune responses to HIV were identified, multiple correlates of immune control have been defined, but which are the cause of a better outcome, and which are the effect, remains elusive. Currently, data support contributions to disease outcome from adaptive immune responses, innate immune responses, host genetic factors, and differences in viral pathogenicity.
immune responses to HIV were identified, multiple correlates of immune control have been defined, but which are the cause of a better outcome, and which are the effect, remains elusive. Currently, data support contributions to disease outcome from adaptive immune responses, innate immune responses, host genetic factors, and differences in viral pathogenicity.
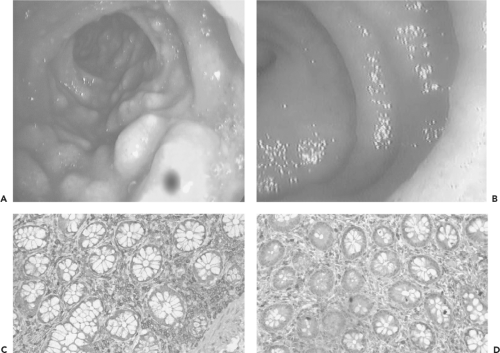 Figure 50.3. Appearance of the terminal ilium in human immunodeficiency virus (HIV) infection. A: Uninfected. B: HIV infected, showing dramatic loss of lymphoid tissue. C: Uninfected, showing numerous CD4 T cells in the lamina propria by immunohistochemical staining. D: HIV infected, showing depletion of CD4 T cells in the lamina propria. |
Innate Immune Responses
The initial immune response to HIV involves innate immune mechanisms. Toll-like receptors (TLRs) on dendritic cells (DCs) are activated by a combination of HIV-1 RNA via TLR7 and microbial products (discussed earlier) via other TLRs, resulting in high levels of interferon alpha (IFN-α), IL-12, tumor necrosis factor alpha (TNF-α), and IL-6, which together result in a profound activation of the immune system.23,42,169 These activated DCs can be infected with HIV, which may further contribute to impaired DC function in infected persons.245,355 Natural killer (NK) cells are also part of the initial innate immune response to HIV, and their numbers are greatly increased in acute infection. Because the increase in NK cells coincides with the initial decline in viremia after acute infection, these cells are thought to have a key role as an antiviral defense mechanism. Moreover, NK cells appear to be largely anergic in chronic infection, suggesting that a pool of nonfunctional NK cells accumulates over time.7 The fact that expression of a killer cell inhibitory receptor, KIR3DS1, and a particular sequence in its ligand human leukocyte antigen (HLA) Bw4, is associated with slower disease progression257 also argues for an important role of these cells in infected persons.
Adaptive Humoral Immune Responses
HIV infection is associated with the development of antibodies that appear within weeks of initial infection. Although most of these antibodies bind, but do not neutralize,319 a subset of these can neutralize virus, either by binding directly to the envelope glycoprotein trimer on the surface of free virions or following CD4-gp120 binding, preventing the fusion of the viral and cell membranes that is essential for viral entry.47 The envelope is heavily glycosylated, and these sugars prevent antibody binding to the underlying peptidic structure. These neutralizing antibody responses nevertheless are sufficiently strong to influence viral evolution; indeed, the virus population in vivo is rapidly replaced by mutant virus that escapes recognition.339,412 This selection is an iterative process: new antibody responses develop to neutralize the mutant virus, which then escapes again. These responses are primarily narrowly directed
and do not appear to be able to broadly cross-neutralize many strains of HIV. During the chronic phase of infection, there is little evidence to indicate an ongoing contribution of neutralizing antibodies to immune control, as shown by depletion of antibody-generating B cells in an animal model of AIDS virus infection.357 Having said this, it is clear that sera from chronically infected individuals can potently neutralize multiple strains of HIV broadly.27,99,235,382,409 Isolation of antibodies from these individuals has proven to be a rich source of monoclonal antibodies with broad and potent activity, and has led to the identification of potential targets of neutralizing antibodies for vaccine development.
and do not appear to be able to broadly cross-neutralize many strains of HIV. During the chronic phase of infection, there is little evidence to indicate an ongoing contribution of neutralizing antibodies to immune control, as shown by depletion of antibody-generating B cells in an animal model of AIDS virus infection.357 Having said this, it is clear that sera from chronically infected individuals can potently neutralize multiple strains of HIV broadly.27,99,235,382,409 Isolation of antibodies from these individuals has proven to be a rich source of monoclonal antibodies with broad and potent activity, and has led to the identification of potential targets of neutralizing antibodies for vaccine development.
Adaptive Cellular Immune Responses
CD8 T-Cell Responses
Adaptive immunity to viruses generally involves the rapid expansion of CD8 T cells that recognize viral proteins on the infected cell surface, presented by HLA antigen class I molecules. Recognition, via the T-cell receptor (TCR), of a foreign (nonself) protein presented as a short 8 to 10 amino acid peptide in the groove of a class I molecule can result in the directed lysis of that cell by CD8 T cells. Depending upon the kinetics of virus production, peptide presentation via class I, CD8 T-cell recognition, and the activation state of the CD8 T cell, virus-infected cells potentially can be eliminated before they are able to produce significant progeny virus.
In the early weeks following HIV infection, viral load is reduced from an average of 1 × 106 copies/mL to an average of 30,000 copies/mL.246,344 Coincident with this drop is the appearance of HIV-specific CD8 T cells, suggesting that these cells have an active antiviral effect36,221; animal model data support this conclusion.194,356 When CD8 T cells are removed by administration of a CD8-specific monoclonal antibody in animals with chronic SIV infection, viral load dramatically increases, and subsequently declines again coincident with the reemergence of these cells. If CD8 T cells are depleted before acute infection in this model then viral load remains at the initially high levels.356 Therefore, it is clear that these cells play an ongoing role in viral containment. Because the latent viral reservoir remains invisible to these responses, CD8 T cells are unable to eradicate HIV.
The breadth and magnitude of CD8 T-cell responses can vary widely among infected persons, as measured by the ability of these cells to secrete IFN-γ following recognition of their cognate epitope. Despite animal model data showing the clear role of these cells in immune containment, other studies have failed to provide strong and consistent evidence that these responses are associated with suppression of the plasma viral load in humans. Responses have been observed to all nine HIV viral proteins and, in most persons, an average of more than a dozen viral peptides are simultaneously targeted.4,25 The total percent of CD8 T cells directed against HIV in the chronic phase of infection can be 25% or higher, and yet, the magnitude of these responses does not correlate with control of viremia.25 One relatively consistent finding is that targeting of HIV-1 Gag over other viral proteins is associated with a lower viral load.326,435 Although initial animal studies suggested that targeting of proteins expressed early in the viral life cycle (e.g., Nef) might be advantageous, this finding has not been consistently borne out in larger studies in humans, although it is clear that Nef is one of the most common viral antigens targeted by the CD8 T-cell response during acute HIV infection.117,146 Data also indicate that the HLA B alleles, as opposed to the A and C alleles, play the dominant role in modulating viral load,213 again suggesting a key role for CD8 T cells in immune containment, but debate continues whether this is cause or effect.
Whereas these data clearly indicate that CTLs are involved in viral control, it is often difficult to determine their exact contribution in human studies. One reason for this difficulty is that both the virus and the CTL response are constantly adapting to each other over the course of the infection. Although each person has up to six different class I alleles to present viral peptides at the cell surface, only a subset of these are used in acute infection.6 That initial immune response is therefore narrowly directed to only a few epitopes, most of which rapidly escape, and other epitopes become targeted as the response broadens over time well after viral set point is reached.9 In addition, it is clear that not all CD8 T cells are the same, and based upon their different phenotypes and functions, they may have profoundly different antiviral effects.121,241
Virus-Specific CD4 T Cells
The optimal production of antibodies and the function of CD8 T cells depends on the presence of virus-specific CD4 T cells.92 The magnitude of these responses, as measured by IFN-γ production, is consistently much lower than the CD8 T-cell responses, and a small number of peptides are recognized by most individuals.25,326 In contrast to the wide spectrum of responses mediated by CD8 T cells, the adaptive CD4 T-cell response is primarily directed against the Gag protein, for reasons that are unclear. In the early stage of acute infection, a robust induction of HIV-specific CD4 T cells occurs, and these cells clearly provide help to CD8 T cells, as evidenced by the ability of CD8 T cells in acute infection to proliferate in vitro in response to cognate epitope recognition.236 The ability of CD4 and CD8 T cells to proliferate in vitro is lost over the first few months of untreated infection, but the acquired defect in CD8 T-cell proliferation can be restored in vitro by the addition of CD4 T cells obtained at the time of acute infection or by IL-2.185,236 Although HIV-specific CD4 T-cell function has been augmented through therapeutic interventions (vaccination, treatment interruptions, and IL-2 therapy), none of these approaches has led to improved virologic or clinical outcomes.3,185,207,340,344,346
CD4 T cells can be classified based on their major functions into multiple lineages, including T helper 1, T helper 2, T helper 17, follicular T helper, and T regulatory. Most HIV-specific CD4 T cells fall within the T helper 1 category based on their secretion of IFN-γ, but their overall function appears to be impaired.161,185 The fact that HIV preferentially infects and depletes HIV-specific CD4 T cells may partially explain the low frequency of HIV-specific CD4 T cells and their inability to adequately coordinate humoral and CD8 T-cell responses to HIV.102 In addition, the phenotype and function of opportunistic pathogen-specific CD4 T cells may affect their susceptibility to infection and depletion by HIV, thereby explaining some of the variation in onset of different opportunistic infections.52,139 Specifically, CD4 T cells that express β chemokines (which block HIV entry by binding to CCR5) are more resistant to HIV infection and depletion than those that do not.52,139
Host Genetics and Viral Control
One of the strongest predictors of disease progression is the HLA type of the host. The HLA class I alleles B*27 and B*57 are associated with low viral load and prolonged asymptomatic
infection,201 although additional alleles have also been shown to affect viral load (Fig. 50.4). Other HLA alleles have been associated with more rapid disease progression, in particular a subtype of the HLA B35 allele referred to as HLA B35 px.133 It is now clear that the HLA B alleles are dominant in terms of having an impact on viral load.213 On a population level, clear evidence indicates viral imprinting by host CD8 T-cell responses: Persons with certain HLA alleles have a significantly increased prevalence of certain viral polymorphisms that appear to be driven by immune escape.208 Animal studies have shown clearly that the major histocompatibility complex (MHC) type can influence both disease progression and the relative contribution of cellular and humoral immune responses to viral control.251 Immunization of humans with candidate HIV vaccines has also shown HLA-related differences in immunogenicity.202 Remaining to be identified are the precise mechanisms whereby these alleles mediate a protective or detrimental effect, or enhance immunogenicity of candidate vaccines, or whether this represents a linkage to some other genetic factor that modulates outcome. In addition to HLA alleles, other genetic factors may also have an impact on disease progression, and are discussed below.
infection,201 although additional alleles have also been shown to affect viral load (Fig. 50.4). Other HLA alleles have been associated with more rapid disease progression, in particular a subtype of the HLA B35 allele referred to as HLA B35 px.133 It is now clear that the HLA B alleles are dominant in terms of having an impact on viral load.213 On a population level, clear evidence indicates viral imprinting by host CD8 T-cell responses: Persons with certain HLA alleles have a significantly increased prevalence of certain viral polymorphisms that appear to be driven by immune escape.208 Animal studies have shown clearly that the major histocompatibility complex (MHC) type can influence both disease progression and the relative contribution of cellular and humoral immune responses to viral control.251 Immunization of humans with candidate HIV vaccines has also shown HLA-related differences in immunogenicity.202 Remaining to be identified are the precise mechanisms whereby these alleles mediate a protective or detrimental effect, or enhance immunogenicity of candidate vaccines, or whether this represents a linkage to some other genetic factor that modulates outcome. In addition to HLA alleles, other genetic factors may also have an impact on disease progression, and are discussed below.
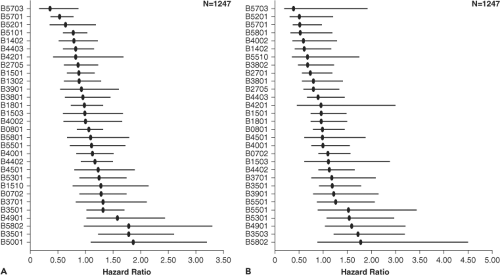 Figure 50.4. Relative hazard of disease progression by human leukocyte antigen (HLA) class I type. A: Relative hazard of progression to a CD4 count of less than 200/mm3. B: Relative hazard of progression to acquired immunodeficiency disease syndrome (AIDS) by the 1987 AIDS defining criteria. |
Reasons for Immune Failure
Functional Defects
Why is it that the immune system is unable to contain HIV replication, despite detection of huge numbers of HIV-specific immune cells? An increasing amount of evidence suggests that the problem is not in the number of antiviral cells but in their function, and that most HIV-specific CD8 T cells have some degree of functional impairment. The list of these potential defects is long, and includes lack of ability to express multiple cytokines simultaneously, to kill infected target cells, or at least to inhibit HIV replication, proliferate, and fully mature.26,91,170,171,272 Which of these aspects of the CD8 T-cell response in chronic HIV infection is a cause as opposed to an effect of ongoing viral replication is often unclear. What is clear, however, is that chronic antigenic stimulation by persistent viruses can lead to exhaustion of CD8 T cells by preventing them from completing the normal progression to renewable memory cells.416,417 In addition, studies in a murine model of chronic viral infection with many similarities to HIV infection in humans suggest that the expression, on CD8 T cells, of molecules associated with T-cell regulation (i.e., programmed death 1 [PD-1], 2B4, CD160, lymphocyte activation gene 3 [LAG-3]) may be affected by the persistent antigenic stimulation of chronic viremia, rendering those CD8 T cells ineffective in clearing virus.21,28 Studies of HIV-specific T cells have generally confirmed the presence of aberrant expression of co-regulatory molecules that can adversely affect HIV-specific CD8 T-cell function and longevity.90,307,396,429 Co-regulatory molecule expression can also be aberrant on CD4 T cells, thereby imparting indirect effects on the CD8 T-cell response.206
Immune Escape
The role of CD8 T cells as an antiviral mechanism is clearly implied by studies showing viral mutations arising within targeted CD8 T-cell epitopes. Studies clearly demonstrate substantial immune selection pressure as evidenced by frequent
polymorphisms linked to specific HLA alleles208 and the accumulation of HLA-associated changes during acute HIV infection. Of note, the effectiveness of CD8 T-cell responses has been linked to the fitness cost associated with viral escape, adding further evidence in support of the fact that HIV-specific CD8 T cells, although functionally impaired, still exert significant antiviral pressure on HIV, even late into infection.79
polymorphisms linked to specific HLA alleles208 and the accumulation of HLA-associated changes during acute HIV infection. Of note, the effectiveness of CD8 T-cell responses has been linked to the fitness cost associated with viral escape, adding further evidence in support of the fact that HIV-specific CD8 T cells, although functionally impaired, still exert significant antiviral pressure on HIV, even late into infection.79
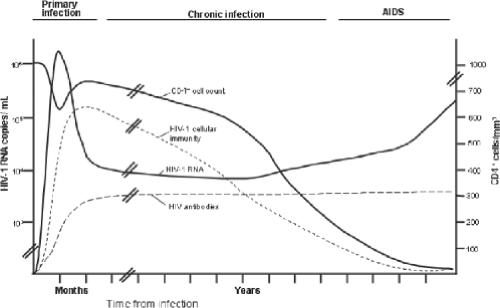 Figure 50.5. Changes in plasma human immunodeficiency virus type 1 (HIV-1) RNA level, CD4+ cell count, HIV-1-specific antibody titers, and HIV-1-specific cellular immune responses over the course of HIV-1 infection. |
Clinical Features
Advancing immunodeficiency caused by the progressive loss of CD4+ T lymphocytes underlies the cardinal clinical manifestations of HIV-1 infection. The myriad opportunistic infections and malignancies characteristic of AIDS are a consequence of the resulting profound defect in cellular immunity. In addition, a number of clinical syndromes are attributable directly to infection of specific organs by HIV-1. The course of HIV-1 disease can be divided into three stages: primary (or acute) infection, chronic (asymptomatic) infection, and advanced disease (AIDS) (Fig. 50.5). The duration of each stage is highly variable and can be altered by antiretroviral therapy. Several systems for classifying the different stages of HIV-1 disease have been developed, the most widely used of which include those developed by the CDC (Table 50.2) and the World Health Organization (WHO).56,426 Although these classification systems are valuable epidemiologic tools, their role in the assessment and clinical management of individual patients has been more limited.
Table 50.2 CDC Classification System for HIV Infection | |||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||
Primary Infection
Acquisition of HIV-1 infection is accompanied by relatively nonspecific symptoms of an acute viral illness in approximately
50% to 70% of infected individuals.198 Symptoms, which usually begin 2 weeks after exposure, frequently include fever, pharyngitis, headache, arthralgias, myalgias, malaise, and weight loss.85,167 A nonpruritic, maculopapular rash on the face and trunk occurs in up to 70% of cases.395 In addition, generalized lymphadenopathy is a frequent finding. Mucocutaneous ulceration and weight loss help distinguish primary HIV-1 infection from other viral syndromes.125,167 Aseptic meningoencephalitis is the most common neurologic manifestation of primary HIV-1 infection.395 Symptoms of primary infection resolve within 3 to 4 weeks in most patients. Persistence of symptoms beyond 8 to 12 weeks, along with a severely depressed CD4+ T-lymphocyte count and high plasma HIV-1 RNA levels may predict more rapid progression of disease.
50% to 70% of infected individuals.198 Symptoms, which usually begin 2 weeks after exposure, frequently include fever, pharyngitis, headache, arthralgias, myalgias, malaise, and weight loss.85,167 A nonpruritic, maculopapular rash on the face and trunk occurs in up to 70% of cases.395 In addition, generalized lymphadenopathy is a frequent finding. Mucocutaneous ulceration and weight loss help distinguish primary HIV-1 infection from other viral syndromes.125,167 Aseptic meningoencephalitis is the most common neurologic manifestation of primary HIV-1 infection.395 Symptoms of primary infection resolve within 3 to 4 weeks in most patients. Persistence of symptoms beyond 8 to 12 weeks, along with a severely depressed CD4+ T-lymphocyte count and high plasma HIV-1 RNA levels may predict more rapid progression of disease.
Laboratory characteristics of primary HIV-1 infection include lymphopenia and a decrease in the absolute CD4+ T-lymphocyte count, usually accompanied by an increase in circulating activated CD8+ T cells.75 Other hematologic abnormalities are unusual, except for mild thrombocytosis. Modest elevations in serum aspartate transaminase and alkaline phosphate may be present, but clinically significant hepatitis is uncommon.395 Plasma HIV-1 RNA titers generally peak 1 week following the onset of symptoms, averaging 106 to 107 copies/mL, and decline to steady-state levels (between 103 and 105 copies/mL) by 2 months after infection.237,246
Chronic Infection
Following resolution of primary infection and establishment of a virologic quasi-steady state,175 a prolonged period of asymptomatic chronic infection ensues. Although most patients remain asymptomatic during much of this phase, ongoing viral replication and CD4 lymphocyte depletion make the term clinical latency inappropriate. The loss of CD4 T cells proceeds at an average rate of 30 to 60 cells/mm3/year, although CD4 cell counts can remain stable for several years before a period of rapid decline.266 A small proportion of patients (<1%) experience progression to AIDS within 1 to 2 years.280,310 Rapid progression may be associated with transmission of syncytium-inducing (SI) variants of HIV-1 (i.e., viruses that use the CXCR4 coreceptor, also known as X4 viruses).64,224,287
Fatigue and lymphadenopathy are noted by many patients during this otherwise asymptomatic phase of HIV-1 infection. Minor clinical events, such as oral hairy leukoplakia (caused by Epstein-Barr virus infection of oral epithelial cells), oral and vaginal candidiasis, herpes zoster, and a variety of other dermatologic conditions may be early signs of clinical progression. With advancing disease, night sweats and weight loss become more common.
A variety of systemic manifestations of HIV-1 infection involving nearly every organ system can occur during the chronic phase of disease. Remission in response to antiretroviral therapy suggests a direct role for HIV-1 in the pathogenesis of these disorders. Dermatologic conditions are particularly common among HIV-1-infected individuals, including seborrheic dermatitis, papular pruritic eruption, and eosinophilic folliculitis.76 Neurologic manifestations include disorders of both the central nervous system (CNS) and peripheral nervous system, in addition to opportunistic infections and malignancies of the CNS. AIDS dementia complex and distal symmetric polyneuropathy, which occur in up to 27% and 35% of patients, respectively, are among the most frequently encountered HIV-related neurologic complications.373,377 Wasting associated with HIV infection remains highly prevalent in resource-limited settings and is an independent predictor of mortality.250 Anorexia, malabsorption, and inappropriate nutrient utilization all contribute to the loss of lean body mass in patients with AIDS.154,220
Other organ system–specific complications of HIV infection include nonspecific interstitial pneumonitis and lymphocytic interstitial pneumonitis, disorders of the gastrointestinal tract, endocrine dysfunction, anemia and neutropenia, immune-mediated thrombocytopenia, and a variety of rheumatologic syndromes. In addition, HIV-associated nephropathy leading to renal insufficiency is particularly common among African Americans and injection drug users.390,422
Advanced Disease
Opportunistic infections and malignancies are rare in HIV-infected persons with CD4 counts above 500 cells/mm3, but increase as the CD4 count declines below this benchmark. Oral candidiasis, pneumococcal infections, tuberculosis, and reactivation of herpes simplex and varicella zoster viruses become more common. The risk of life-threatening complications, including Pneumocystis jirovecii (formerly, P. carinii) pneumonia, candida esophagitis, disseminated histoplasmosis and other systemic fungal infections, toxoplasma encephalitis, and cryptococcal meningitis increases substantially once the CD4 lymphocyte count drops below 200 cells/mm3.258 Opportunistic infections, such as disseminated Mycobacterium avium complex infection; reactivation of cytomegalovirus (CMV) infection, cryptosporidiosis, and microsporidiosis; and progressive multifocal leukoencephalopathy caused by JC virus reactivation, are all indicative of a profound defect in cellular immunity and usually occur at CD4 counts below 50 cells/mm3. Malignancies associated with AIDS generally are related to underlying viral infection, including Kaposi sarcoma (KS) caused by infection with human herpes virus 8; lymphomas associated with Epstein-Barr virus infection; and cervical and anal carcinoma associated with human papilloma virus infection. Although potent antiretroviral therapy has clearly reduced the risk of KS and non-Hodgkin lymphoma, the effect on Hodgkin lymphoma is less clear.32,33,370 Whereas the incidence of lung cancer is also increased in persons with HIV infection, age-adjusted rates of other malignancies (e.g., breast and prostate cancer) are comparable to those found in the general population.152,369
Non-AIDS Complications of HIV Infection
A growing number of end-organ complications not traditionally considered “AIDS-defining” events have been recognized to occur more frequently in HIV-1–infected patients.253,286 These include an increased risk of cardiovascular disease, non-AIDS defining malignancies, HIV-associated neurologic dysfunction (HAND), and HIV-associated nephropathy (HIVAN), and may also include loss of bone mineral density (BMD) and other changes typically associated with increasing age, suggesting that HIV-1 infection may accelerate the aging process more generally.
With respect to cardiovascular risk, a number of studies suggest increased risk of myocardial infarction in HIV-1–infected patients as compared to control populations matched for traditional cardiovascular risk factors such as age, family history, hyperlipidemia, hypertension, diabetes, and smoking.177,286,398 More striking is the finding that interrupting antiretroviral
therapy significantly increases the risk of cardiovascular events, independent of CD4 cell count.106,313 This increased risk was correlated with an increase in plasma levels of inflammatory markers such as IL-6 and high-specificity C-reactive protein (hsCRP).223,347 In addition, a number of studies have shown adverse changes in surrogate markers associated with cardiovascular risk such as flow-mediated vasodilatation, carotid intima-media thickness, and coronary artery calcification in patients with HIV-1 infection.82,153 The extent to which these changes can be prevented or reversed by antiretroviral therapy is an active area of current research.
therapy significantly increases the risk of cardiovascular events, independent of CD4 cell count.106,313 This increased risk was correlated with an increase in plasma levels of inflammatory markers such as IL-6 and high-specificity C-reactive protein (hsCRP).223,347 In addition, a number of studies have shown adverse changes in surrogate markers associated with cardiovascular risk such as flow-mediated vasodilatation, carotid intima-media thickness, and coronary artery calcification in patients with HIV-1 infection.82,153 The extent to which these changes can be prevented or reversed by antiretroviral therapy is an active area of current research.
Patients with HIV-1 infection also show lower BMD and an increased risk of fractures compared to age-matched, uninfected controls.397 The causes of HIV-associated loss of BMD are poorly understood and most likely are multifactorial, including increased immune activation and inflammation, renal tubular dysfunction, low vitamin D levels, and other endocrine abnormalities.10 Whether vitamin D levels should be monitored in all HIV-infected patients, and at what level to offer supplementation, remains an area of controversy.
Determinants of Disease Progression
Numerous viral and host factors contribute to determining the rate of HIV disease progression. The plasma HIV-1 RNA level, or viral load, reflects the rate of virus replication and is a powerful independent prognostic factor for the risk of disease progression.267,304 Data from the MACS show that in the absence of antiretroviral therapy individuals with plasma HIV-1, RNA levels greater than 100,000 copies/mL 6 months after seroconversion are 10 times more likely to progress to AIDS within 5 years than are those with lower steady-state levels of viremia.265 Similarly, for patients with established HIV-1 infection, a steady-state viral load of more than 30,000 copies/mL is associated with a more than fivefold greater risk of disease progression within 3 years as compared with patients with viral loads of 3,000 to 10,000 copies/mL.266
According to a Poisson regression model based on data from the Concerted Action on Seroconversion to AIDS and Death in Europe (CASCADE) collaboration of 22 cohorts from Europe, Canada, and Australia, in the absence of combination antiretroviral therapy the 6-month risk of AIDS for a 25-year old patient with a CD4 cell count of 350/mm3 ranges from 0.6% at a viral load of 3,000 copies/mL to 2.5% at a viral load of 300,000 copies/mL.311 At a CD4 count of 100 cells/mm3, the AIDS risk at the same viral loads increases to 3.7% and 14.5%, respectively. The risk of disease progression has been reduced substantially since the introduction of potent combination antiretroviral therapy.105 Progression to AIDS among persons infected with HIV-2 proceeds at a significantly slower rate as compared with infection with HIV-1.188,260 Epidemiologic and cohort studies show that HIV-2 infection generally results in lower steady-state levels of viremia (103 copies/mL) and a more gradual decline in CD4 cell counts.15,24,188
Age at the time of infection is an independent risk factor for disease progression, with older persons being at significantly greater risk, perhaps as a consequence of diminished thymic reserve.89,309 The role of gender in determining disease progression is less clear. Most studies show that HIV-1–infected men and women progress to AIDS at similar rates.134,273 Early in the course of infection, however, women tend to have significantly lower plasma HIV-1 RNA levels men.11,12,111 This difference disappears as disease progresses. Because overall progression rates are comparable between the sexes, these observations imply that compared with men, women experience HIV-1 disease progression at lower viral loads. Whether to initiate antiretroviral therapy at lower levels of viremia in infected women than in men remains controversial (see below).
Although virus replication, and consequently viral load, is the engine that drives progression to AIDS, the CD4 cell count is the most useful marker for predicting the immediate risk of developing particular opportunistic infections.105 Moreover, differences in viral load explain only a small fraction of the variability in rates of CD4 cell decline in patients not receiving antiretroviral therapy,343 suggesting that other factors such as immune activation triggered by translocation of microbial products across a damaged intestinal mucosa drive CD4 cell loss in HIV infection (see Pathogenesis). Indeed, the proportion of activated CD8+ T cells, measured as the percentage of cells expressing CD38, predicts the risk of disease progression independently of viral load and CD4 count.31,142,143,240 Levels of IL-6 and hsCRP are elevated in patients with HIV infection and independently predict the development of opportunistic diseases.342
The role of chemokine receptor tropism in determining the rate of disease progression remains unresolved. The prevalence of X4 variants increases with decreasing CD4 cell count, and several studies show a significantly increased risk of disease progression among patients with X4 (SI) virus.45,217,278,362 Macaques infected with a simian-HIV (SHIV) (SIV/HIV chimera) that expresses an X4 HIV-1 envelope show rapid depletion of CD4 cells, suggesting a causal role of X4 viruses in rapid disease progression.178,288 X4 variants, however, emerge in only half of patients who progress to AIDS.38,336 The long interval between infection and emergence of X4 viruses in most patients argues for strong selection against X4 viruses early in the course of HIV disease. Therefore, the possibility that emergence of X4 variants is a consequence, rather than a cause, of advancing immunodeficiency remains a plausible alternative explanation for the apparent association of X4 virus with disease progression. The development of chemokine receptor antagonists as a novel class of antiretroviral agents may provide new tools to address this important pathogenesis question.
The risk of disease progression is moderated also by a variety of host genetic factors. Among the most important are polymorphisms in the genes encoding the chemokine co-receptors and their ligands, and in the HLA genes. HIV-1 uses one of two chemokine receptors as co-receptors for virus entry into CD4 cells: CCR5 or CXCR4.97,103,113 Approximately 10% of Caucasians carry a defective allele that has a 32-base pair (bp) deletion in the CCR5 gene (ccr5Δ32); 1% are homozygous for this deletion and resist infection by R5 viruses.239,352 Infected individuals who are heterozygous for the ccr5Δ32 allele have a slower rate of disease progression than do those who are homozygous for the wild-type allele.94 This effect is limited to patients with R5 strains of HIV-1, presumably because syncytium-inducing viruses use the CXCR4 receptor.40,270 A mutation in the CCR2 gene, CCR2–64I, likewise reduces the rate of disease progression,219,376 possibly by delaying emergence of X4 virus.403 By contrast, mutations in the promoter region of CCR5 (CCR5 P1) are associated with accelerated disease progression.256 Other analyses have failed, however, to show a significant effect of CCR5 and CCR2 genotype after
controlling for CD4 cell count, plasma HIV-1 RNA level, and viral tropism.141,184
controlling for CD4 cell count, plasma HIV-1 RNA level, and viral tropism.141,184
Copy number of the gene encoding the natural ligand for CCR5 (CCL3L1, previously known as MIP-1α) also influences the rate of disease progression. Higher CCL3L1 copy number is associated with a lower risk of progression, which is most pronounced in patients with polymorphisms that reduce the level of functional CCR5 on the cell surface.145 Variable effects on progression have been noted for the SDF1-3′A polymorphism, which affects the untranslated region of the mRNA encoding stromal-derived factor (SDF-1, also known as CXCL12), the natural ligand of CXCR4.86,145,421
Several studies have related host HLA haplotype to the rate of HIV disease progression. For example, presence of the HLA-B27 and B57 alleles is associated with slow progression.8,112,305 Conversely, patients carrying class I alleles B*35 or Cw*04 progress to AIDS significantly more rapidly than do those lacking these alleles.51,133 Maximal heterozygosity at HLA class I loci A, B, and C is associated with delayed onset of AIDS.201 Given that HLA antigen class I molecules play an essential role in antigen presentation to CD8+ CTL, these findings provide strong, albeit indirect, evidence for the importance of CTL in moderating the rate of HIV disease progression.
Variation in the killer immunoglobulin-like receptor (KIR) genes also affects the course of disease. These receptors are found on NK cells and regulate NK activity by recognition of certain HLA class I molecules on the surface of target cells.404 The effect of KIR alleles appears to be related to the presence or absence of specific HLA-B or HLA-C alleles.137 For example, when present together with the HLA-B Bw4-80Ile allele, the activating KIR allele KIR3DS1 is associated with delayed disease progression.257 By contrast, in the absence of HLA-B Bw4-80Ile, the presence of KIR3DS1 is associated with more rapid disease progression. Similarly, presence of the inhibitory KIR allele KIR3DL1 together with HLA-B*57S is highly protective against disease progression.244 The KIR2DS2 allele appears to be associated with more rapid CD4 decline over time, but has no effect on viral load, whereas HLA-B*5701 and B*2705 alleles are associated with significantly lower levels of viremia.136
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


