Hemophagocytic Lymphohistiocytosis
Sa A. Wang, MD
Key Facts
Terminology
Cytokine dysfunction, resulting in activation of T lymphocytes and macrophages
Leads to systemic symptoms and organ damage
Etiology/Pathogenesis
Inherited genetic or acquired defect leading to depressed NK- &/or cytotoxic T-cell activity
Defect in packaging, exocytosis, or function of cytotoxic granules
Microscopic Pathology
In lymph nodes
Sinuses are infiltrated by bland histiocytes showing phagocytic activity
Underlying etiology may coexist in same lymph node
Top Differential Diagnoses
Rosai-Dorfman disease
Histiocytic sarcoma
Langerhans cell histiocytosis
Diagnostic Checklist
Clinical and laboratory criteria (5 out of 8 criteria)
Fever
Cytopenias
Splenomegaly
Hypertriglyceridemia &/or hypofibrinogenemia
Serum ferritin > 500 µg/L
Hemophagocytosis
Low or absent NK-cell activity
Soluble CD25 (sIL-2 receptor) > 2,400 U/mL
HLH diagnosed by molecular testing
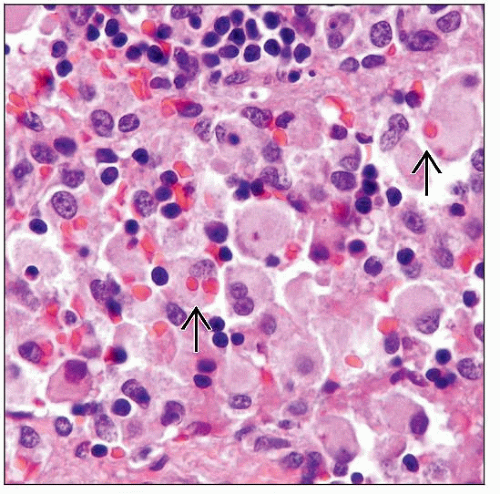 In a lymph node involved by hemophagocytic lymphohistiocytosis (HLH), the sinus is expanded by numerous mature histiocytes/macrophages, some exhibiting phagocytosis of mostly erythrocytes  . . |
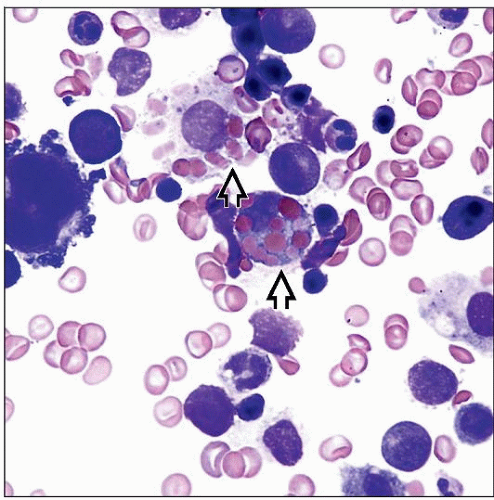 This patient presented with fever and pancytopenia prompting bone marrow examination. Bone marrow aspirate smear (Wright-Giemsa stain) shows erythrophagocytosis 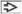 . . |
TERMINOLOGY
Abbreviations
Hemophagocytic lymphohistiocytosis (HLH)
Synonyms
Hemophagocytic syndrome
Erythrophagocytic lymphohistiocytosis
Viral-associated hemophagocytic syndrome
Definitions
Cytokine dysfunction, either due to inherited or secondary causes
Results in overwhelming activation of normal T lymphocytes and macrophages
Leads to systemic symptoms and organ damage
ETIOLOGY/PATHOGENESIS
Inherited vs. Secondary/Acquired Defects
Inherited genetic defects
Result in depressed functional cytotoxicity of natural killer (NK) cells and cytotoxic T cells
T cells cannot terminate the stimulatory responses they receive from antigen-presenting cells
NK cells and cytotoxic T cells cannot clear infection
Perforin (PRF1) mutation
Localized at 10q21-22
Results in decrease or absence of perforin in NK or CD8(+) T cells
Griscelli syndrome (MUNC13-4 defect)
Lack of protein rab27a, which controls secretion of lytic granules in NK and cytotoxic T cells
Chediak-Higashi syndrome (LYST gene defect)
Cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) is involved
Failure to move CTLA-4 from secretory lysozymes to cell membrane
Syntaxin gene mutations
NK cells fail to degranulate when encountering susceptible targets
X-linked lymphoproliferative disease (XLP)
SH2D1A gene mutation
Defect in T-cell signal transduction
T-cell lytic defect against EBV-infected B lymphocytes
Trigger vigorous cytotoxic cellular responses
a.k.a. Duncan syndrome
Defect in apoptosis-caspase 3 leading to accumulation of T cells
Acquired/secondary defects
It is not clear how NK-cell and T-cell function is impaired
Many infectious agents are reported to be associated with HLH
Viruses
EBV is most common, CMV, parvovirus B19, herpes simplex
Herpes varicella-zoster, measles, human herpesvirus 8, HIV
Adenovirus, respiratory syncytial virus, parainfluenza virus, enteroviruses
Bacteria
Pseudomonas aeruginosa, staphylococci, streptococci
Escherichia coli and Brucella abortus
Mycobacteria: Mycobacterium tuberculosis
Parasites: Leishmania donovani; Plasmodium species
Fungi
Histoplasma capsulatum, Penicillium marneffei, Aspergillus
Cryptococcal meningitis, histoplasmosis, and disseminated Trichosporon beigelii
Autoimmune disorders
Systemic lupus erythematosus, rheumatoid arthritis
Still disease, polyarteritis nodosa
Mixed connective tissue disease, systemic sclerosis, Sjögren syndrome
Malignancies associated with HLH
T- and NK-cell lymphomas
Acute myeloid leukemias and myelodysplastic syndromes
Acute lymphoblastic leukemia/lymphoma (B or T cell)
B-cell lymphomas; carcinomas
Other diseases reported associated with HLH
Post-transplant
Pulmonary sarcoidosis
Common Etiologies of Inherited & Secondary/Acquired HLH
Inappropriate immune reaction caused by
Proliferation &/or activation of T cells either due to genetic defects or secondary causes
Defect in packaging, exocytosis, or function of cytotoxic granules
Production of large quantities of cytokines including
Interferon-γ, TNF-α, and granulocyte-macrophage colony-stimulating factor
Interleukin-1 (IL-1) and interleukin-6 (IL-6)
Associated with macrophage activation
Inadequate apoptosis of immunogenic cells
CLINICAL ISSUES
Epidemiology
Incidence
1.2 children per million per year or 1 case per every 50,000 births
Incidence in adults is unknown
Age
Familial form frequently affects infants
Birth to age 18 months most common (70-80%)
Rare familial cases can affect adolescents and adults
Acquired form can occur at any age
Gender
M:F = 1:1
Ethnicity
No predilection for any race
Presentation
Fever: ≥ 7 days of fever as high as 38.5°C (101.3°F)
Easy bruisability and pallor related to cytopenia(s) or coagulopathy
Splenomegaly: Spleen palpable > 3 cm below costal margin
Central nervous system symptoms
Seizures, ataxia, hemiplegia, mental status changes, irritability
Skin rash: Scaly and waxy lesions; rashes on scalp and behind ear
Lymphadenopathy
Hepatomegaly, jaundice
Pleural effusion; ascites
HLH occurring in the context of malignancies
May mask underlying malignancies
Often associated with severe infection due to intrinsic or iatrogenic immunocompromised state
Laboratory Tests
Cytopenia(s), often pancytopenia
Hemoglobin < 9.0 g/dL
Platelets < 100,000/µL
Absolute neutrophil count < 1,000/µL
Hypofibrinogenemia
Fibrinogen < 1.5 g/L, or > 3 standard deviations (SD) below normal value for age
Hypertriglyceridemia
Fasting triglycerides ≥ 2.0 mmol/L, or > 3 SD above normal value for age
Increased serum ferritin
> 500 µg/L
Glycosylated ferritin < 20% of total ferritin
Levels parallel to course of disease
Can use to monitor disease activity
80% specific for diagnosis of HLH
Abnormal liver function
Hyperbilirubinemia
Hypoalbuminemia
Increased aspartate aminotransferase (AST) and alanine aminotransferase (ALT) levels
Serum lactate dehydrogenase (LDH) increased
Defect in NK-cell activity; adequate NK-cell number
Can differentiate between the HLH subtypes
Increased concentrations of circulating soluble interleukin receptor (sIL-2R)
Molecular diagnosis
Gene mutation analysis
Treatment
HLH-2004 protocol recommended by Histiocytosis Association of America
8-week period with dexamethasone, etoposide, and cyclosporine
Resolved nonfamilial HLH does not require continuation of therapeutic regimen
Children with persistent nonfamilial disease or familial disease should continue therapy with cyclosporine, plus etoposide and dexamethasone pulses, until stem cell transplant
Intrathecal methotrexate is used for persistently abnormal CSF or progressive neurologic symptoms
Stem cell transplantation for patients with
Familial HLH
Children and adults with persistent nonfamilial disease
Prognosis
With HLH-2004 protocol
3-year probability of survival was 51% for verified familial cases
55% for entire group of HLH patients
Stem cell transplant
Matched transplant: Long-term disease-free rate ˜ 70%
Special Form of HLH-Macrophage Activation Syndrome (MAS)
Occurs in children and adults with autoimmune diseases, especially
Systemic-onset juvenile rheumatoid arthritis (or systemic juvenile idiopathic arthritis): 10-50% of patients
Adult-onset Still disease
Lupus erythematosus
Kawasaki disease
Different rheumatic conditions exploit different pathways to arrive at an MAS phenotype
Clinical and laboratory features
Share many characteristics with HLH
Defective NK-cell function and low perforin expression
Clinical signs and symptoms similar to HLH
Hemophagocytosis is present in bone marrow, spleen, lymph node
High ferritin levels
Features that differ from HLH
Less severe cytopenias
Not associated with severe infection
More severe cardiac impairment and more pronounced coagulopathy
Very high C-reactive protein level
Therapy
Cyclosporine and steroids
If these do not work, use HLH-2004 protocol
IMAGE FINDINGS
General Features
No specific imaging patterns are diagnostic of HLH
CT or ultrasonography findings
Ascites, pleural effusion
Gallbladder wall thickening
Lymphadenopathy
MR for CNS involvement
MICROSCOPIC PATHOLOGY
Histologic Features
Proliferation of small mature histiocytes
Histiocytes show phagocytic activities
Tissue infiltrate and cellular injury
Increased lymphocytes; mostly T cells
Cytologic Features
Histiocytes are mature and cytologically benign
Have vesicular nuclei and abundant cytoplasm
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


