Sequence of events in normal hemostasis. (With permission from O’Leary JP, Tabuenca A, eds. Physiologic Basis of Surgery. 4th ed. Philadelphia, PA: Wolters Kluwer Health/Lippincott Williams & Wilkins; 2007.)
Normal Hemostasis
•Injury to vessel endothelium exposes collagen, which stimulates platelets
•Thrombin generation is also a major catalyst for platelet activation
•Vasoconstriction is endothelium-initiated
•Platelet adhesion is mediated by binding of platelet surface receptor GPIb to von Willebrand factor (vWF) within exposed collagen
•Platelet aggregation occurs when fibrinogen acts as a bridge between activated platelets via GPIIb/IIIa receptors
•Platelets then release granules containing ADP, serotonin, and arachidonic acid which is converted to thromboxane A2 by cyclooxygenase (COX)
•Thromboxane A2 stimulates further vasoconstriction and platelet aggregation

•Factor VIII-vWF is the only factor not made in the liver (made by endothelium)
Aspirin irreversibly inhibits the cyclooxygenase enzyme, which inhibits platelet aggregation (effect lasts up to 10 days).
The endothelium is an important factor in maintaining blood fluidity and preventing thrombosis in the face of vascular insults. What is the most potent endothelial factor responsible for preventing intravascular thrombosis?
Nitric oxide. Other factors include prostacyclin, thrombomodulin, heparin sulfate proteoglycan, and tissue plasminogen activator.
The Endothelium
•To prevent platelet aggregation, endothelium generates both nitric oxide (NO) and prostacyclin (PGI2)
•Natural heparin sulfate proteoglycan serves as a cofactor for antithrombin III, preventing thrombin formation
•Factors that disrupt the normal endothelial function (endotoxin, tumor necrosis factor alpha, interleukin-1, and hypoxemia) can lead to prothrombotic states
A 43-year-old man was diagnosed to have von Willebrand disease after prolonged bleeding following a tooth extraction. He now presents for an elective laparoscopic cholecystectomy. What measures should be performed to help prevent abnormal bleeding?
Desmopressin (1-deamine-8D arginine vasopressin, DDAVP) should be given prior to surgery to boost levels of vWF and Factor VIII. If unresponsive to DDAVP, factor VIII-vWF concentrates are available.
von Willebrand Disease
•Most common congenital bleeding disorder (1% prevalence)
•Characterized by a deficiency or dysfunction of von Willebrand factor (vWF)
•Stored in endothelial cells and megakaryocytes
•Assists in platelet adhesion
•Carrier protein for factor VIII
•Links GPIB receptors on platelets to collagen to cause platelet adhesion at the bleeding site
•Type I: Quantitative decrease in vWF
•Type II: Qualitative decrease in vWF
•Type IIa—abnormally small vWF (large multimers needed for platelet binding)
•Type IIb—rapid clearance of large vWF multimers
•Type III: Near absence of vWF
•PT is normal, aPTT normal/abnormal, bleeding time prolonged, ristocetin test is positive
•Presents with mucosal bleeding, petechiae, epistaxis, and menorrhagia
•Acquired forms of vWD (secondary to vWF directed antibodies) occur in lymphoproliferative disorders, malignancy, drugs, hypothyroidism, and autoimmune diseases
•Treatment
•Desmopressin (DDAVP) causes release of endogenous stores of vWF from Weibel-Palade bodies in endothelium
•Raises levels of factor VIII
•Can induce flushing, tachycardia, and headaches due to vasoactive effects
•Is not effective for all patients
•Factor VIII-vWF concentrates
•Cryoprecipitate may also be used if Factor VIII-vWF concentrates are not available
•Aminocaproic acid and tranexamic acid can be administered as a mouthwash for dental procedures
A 25-year-old man is traveling through South America and becomes severely ill with malaria. He is given quinine and improves from his malarial infection. However, a few days later, he develops thrombocytopenia, anemia, altered mental status, and renal failure. What does he have and how can it be treated?
He has thrombotic thrombocytopenic purpura (TTP), which is most often idiopathic, but can be caused by quinines, some cancers, and immunosuppression.
Thrombotic Thrombocytopenic Purpura
•Caused by an inability to degrade large vWF multimers due to deficiency in protease ADAMTS13
•Signs: thrombocytopenia, hemolytic anemia, altered mental status, renal failure, and fever
•Death is most commonly secondary to intracerebral hemorrhage or acute renal failure
•Treatment
•Large volume plasmapheresis
•Splenectomy is rarely indicated
Idiopathic Thrombocytopenic Purpura
•Caused by autoantibiodies (IgG) to platelets → platelet sequestration in the spleen and destruction
•Signs: Petechiae and bleeding gums
•Causes: Idiopathic, HIV, HCV, SLE
•Treatment: Steroids, IVIG, splenectomy
•Splenectomy removes the source of IgG production and phagocytosis
Uremic Bleeding
•Platelet dysfunction due to decreased adhesion and aggregation mostly from altered GPIIb/IIIa
•Also have abnormal expression of GPIb receptors, vWF and altered release of ADP, serotonin, and thromboxane A2
•Treatment: Dialysis, DDAVP
•Defective glycoprotein IIb-IIIa receptors
•Prevents platelet aggregation with fibrinogen
•Autosomal recessive
•Signs: Petechiae, bleeding gums, prolonged bleeding time
•Treatment: Platelet transfusion
Bernard-Soulier Syndrome
•Abnormality in the glycoprotein Ib (vWF receptor)-Factor V-Factor IX complex
•Results in abnormal platelet adhesion
•Autosomal recessive
•Signs: Petechiae, bleeding gums, prolonged bleeding time
•Treatment: Platelet transfusion
A 5 year-old boy with a history of significant bleeding after small cuts is playing outside and comes in complaining of severe right knee pain. He states that he was just playing and did not fall on or injure the knee. On further workup, he is found to have hemorrhage into his right knee joint. What further hematologic workup should he have?
His story is consistent with hemophilia A or B and he should have mixing studies, which will normalize the factor deficiency if this is the diagnosis.
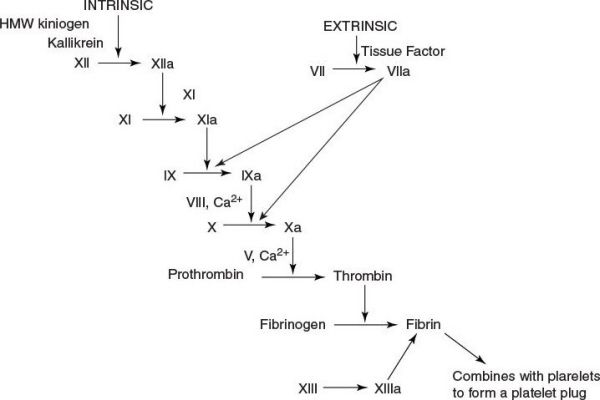
Coagulation cascade.
Hemophilia A (Inherited Factor VIII Deficiency)
•X-linked recessive (primarily affects men)
•Symptoms: Excessive bleeding, spontaneous hemarthroses
•Prolonged aPTT, normal PT, normal bleeding time
•Confirm with mixing studies, which should normalize the factor deficiency
•Factor VIII crosses the placenta and therefore newborns do not bleed at circumcision
•May use DDAVP to raise levels of factor VIII for simple surgical procedures
•Treat with recombinant factor VIII or FFP for severe cases
•Do not aspirate a hemophiliac joint → use ice, keep joints mobile with physical therapy, and give factor VIII and cryoprecipitate
•Goal factor levels of 20% to 30% for minor surgery, 50% to 80% for major surgery
Hemophilia B (Inherited Factor IX Deficiency) “Christmas Disease”
•X-linked recessive
•Clinically indistinguishable from type A
•Laboratory findings similar to type A
•DDAVP not effective
•Treat with recombinant factor IX, FFP, or prothrombin complex concentrate
Inhibitors of the Coagulation Cascade
•Thrombomodulin
•Binds and changes the shape of thrombin
•Becomes a potent activator of protein C and S (anticoagulant)
•Activates thrombin-activated fibrinolysis inhibitor (TAFI), which inhibits fibrinolysis
•Protein C and S
•Potent inhibitors of factor VIII & V
•Antithrombin III
•Does not require activation
•Binds to thrombin
•Inhibits factors VIIa, IXa, Xa, XIa, and plasmin
•Plasmin
•Converted from plasminogen by tissue plasminogen activator (released from endothelium)
•
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


