Heart Failure and Dysrhythmias
Common Sequelae of Cardiac Diseases
Shann D. Kim and Jacquelyn L. Banasik
Key Questions
• What are the common predisposing factors for development of heart failure?
• How do the compensatory responses triggered in heart failure work to restore cardiac output?
• What are the clinical manifestations of heart failure?
• What are the characteristic electrocardiographic features of the common cardiac dysrhythmias?
• What is the clinical significance and usual treatment of each of the common cardiac dysrhythmias?

http://evolve.elsevier.com/Copstead/
Heart failure (HF) and cardiac dysrhythmias (arrhythmias) may occur in association with cardiac diseases from a number of different causes. Heart failure refers to the inability of the heart to maintain sufficient cardiac output to optimally meet metabolic demands of tissues and organs, and is the end stage of most cardiac diseases. Heart failure involves multiple organ systems and is a progressive syndrome. If the contracting ability of the heart is impaired, then blood flow to the systemic circulation will be reduced, and congestion of blood can occur in the pulmonary venous circulation. In patients with HF, these symptoms of fluid overload are described as congestive heart failure (CHF), a term used as a diagnosis in some cases. The term CHF may also be used to differentiate chronic heart failure from acute heart failure. This chapter includes the chronic forms of heart failure; acute heart failure is discussed in Chapter 20 because it commonly results in cardiogenic shock. Disturbances in electrical activity of the heart may signify underlying pathophysiologic processes and may also lead to insufficient cardiac output. Neither heart failure nor dysrhythmia is a primary cardiac disease; therefore, underlying pathophysiologic processes must be investigated.
Heart failure is the fastest-growing cardiac disorder; it affects about 5.7 million Americans. More than 550,000 new cases are diagnosed in the United States each year, with an incidence of 10 per 1000 population after age 65.1 Heart failure is the most common reason for hospitalization in patients older than 65 years.2 The increasing incidence and hospitalization rates of HF reflect aging of the U.S. population, as well as better treatment and an improved survival rate after myocardial infarction (MI).1,3
Heart Failure
Pathogenesis and Diagnosis
A large number of cardiac disorders, including most of those discussed in Chapter 18, can lead to the development of heart failure. Coronary artery disease (CAD) is responsible for 60% to 75% of HF cases, and hypertension (HTN) contributes to nearly 75% of HF cases. CAD contributes to HF progression through mechanisms that include endothelial dysfunction, ischemia, and infarction. CAD and HTN interact to increase the risk of HF.1 In recent years, the impact of obesity and type 2 diabetes mellitus (T2DM) has become important. T2DM is known to accelerate atherosclerosis, and is frequently associated with HTN. Less common causes of HF include dilated cardiomyopathy, congenital heart defects, valvular disorders, respiratory diseases, anemia, and hyperthyroidism. The diagnosis of heart failure is based on the presence of a constellation of signs and symptoms that are characteristic of the syndrome. However, different sets of criteria are in use, including the Framingham Criteria and Minnesota Heart Failure Criteria.4 Commonly used criteria for identifying heart failure include the presence of dyspnea, pulmonary rales, cardiomegaly, pulmonary edema, S3 heart sound, and tachycardia (greater than 120 beats/min), although many other criteria may be applied. No single diagnostic test is available for HF. The diagnosis should be based on a thorough medical history and physical examination.1
CHF is generally classified as systolic or diastolic heart failure.5,6,7 Regardless of specific cause, the pathophysiologic state of heart failure results from the impaired ability of myocardial fibers to contract (systolic failure), relax (diastolic failure), or both.7 Until the late 1980s, systolic dysfunction was thought to be the primary problem in all forms of HF. However, epidemiologic studies have shown that about half of patients with chronic HF have preserved systolic function, but impaired diastolic function.8,9 The severity and differentiation of heart failure patients with systolic failure or preserved systolic function is based primarily on the left ventricular ejection fraction (EF). Ejection fraction is calculated by dividing stroke volume by end-diastolic volume. A normal EF is 60% to 80%. Patients with systolic failure have characteristically low ejection fractions (<40%). Patients with EF greater than 50% do not have significant systolic dysfunction and are categorized as HF with normal EF (HFnlEF). The majority of patients diagnosed with heart failure who have an EF greater than 50% have diastolic dysfunction.8,9 Many patients with low EF also have impaired diastolic function. In general, patients with low EF but no congestive symptoms have a survival rate about the same as those with preserved EF who have congestive symptoms. The highest mortality occurs in patients with both low EF and congestive symptoms.
The overall mortality for heart failure is high with about 50% (42% to 65% in various studies) of patients dying within 5 years of diagnosis.2,5,6 Survival rates vary significantly between genders, with men having a 35% survival rate at 5 years and women having a 53% survival rate. The median survival time after diagnosis is 1.7 years in men and 3.2 years in women.6
Systolic Dysfunction
Patients with systolic dysfunction have reduced myocardial contractility evidenced by a low EF and a reduced dP/dt during ventricular systole. The dP/dt is a measure of inotropy—how quickly the ventricle can develop a forceful contraction. The nature of the impaired contractility is only partially understood; however, myocyte loss, mechanical derangements of myocardial cells, and dysregulation of neurohormones are believed to be critical elements.10
Impaired contractility attributable to myocardial infarction (MI) is a common cause of heart failure.11 MI, with cell death and loss of contractile elements, reduces the heart’s contractile force. The degree of pump failure is related to the amount of heart muscle lost. In patients with heart failure, myocardial cells are also subject to high rates of apoptosis or programmed cell death. Apoptosis can be triggered by excessive stimulation by certain neurohormones and by ischemia. Over time, the loss of myocardial cells contributes to reduced contractility. In severe systolic HF, the EF may fall below 15% or 20%. In general, the prognosis worsens as EF decreases. Symptomatic patients with systolic heart failure often have impaired diastolic function, which is associated with a higher mortality rate.7
Inadequate supplies of oxygen to the contracting cells may impair contractility because each myosin cross-bridge cycle requires a molecule of adenosine triphosphate (ATP). When ATP production is low, fewer cross-bridge cycles are completed with each contraction, which results in a reduced EF.
β1-Receptor down-regulation is thought to be an important mechanism of impaired systolic function. Chronic overexcitation of cardiac β1 receptors by sympathetic neurotransmitters (e.g., norepinephrine) leads to a reduction in the number of β1 receptors, and results in a myocardium that is less responsive to sympathetic stimulation and adrenergic drug therapy. β1-Receptor blocking agents have been shown to improve EF and also to reduce mortality, which lend support to the view that chronic excessive sympathetic nervous system (SNS) activation is detrimental to cardiac function.12–14
Diastolic Dysfunction
Coronary artery disease and hypertension are the two main causes of diastolic dysfunction, just as they are the primary causes of systolic failure. Why the same disease processes result in different cardiac dynamics in different individuals is not known. Some patients have isolated systolic failure, whereas others have isolated diastolic failure. Some patients exhibit both abnormal systolic and diastolic function. HF with normal EF is more common in women, the elderly, and those with no history of MI.7 Diastolic failure is a disorder of myocardial relaxation. In this condition, the left ventricle is excessively noncompliant and does not fill effectively. Two separate functional processes occur during the diastolic relaxation phase: The first is an energy-requiring process (lusitropy) that removes free calcium ions from the cytoplasm by pumping them back into the sarcoplasmic reticulum and across the cell membrane into the extracellular fluid. Removal of calcium ions inhibits cross-bridge formation and allows the thick and thin filaments of the sarcomere to passively slide apart. Ischemia, with subsequent ATP deficiency, interferes with the efficiency of calcium ion removal and can impair the active phase of diastolic relaxation.
The second process is passive stretch of the ventricular myocardium to accommodate filling. Passive compliance of the ventricle can be decreased by deposition of fibrin and collagen during scar formation or by hypertrophic thickening of the ventricular wall. Both active and passive processes may be impaired together and are difficult to distinguish clinically.
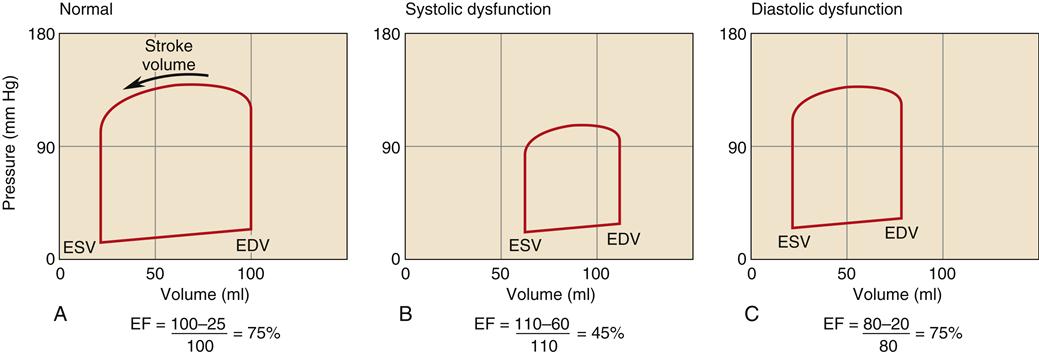
The hallmark of HF with normal EF is that the patient exhibits clinical manifestations of HF, including low cardiac output, pulmonary congestion, and edema formation, but has a normal EF (usually defined as greater than 50%), indicating absence of significant systolic impairment.8,9 Because prognosis and treatment recommendations may differ, an echocardiogram to measure EF is recommended in all patients with HF to determine the presence of isolated diastolic dysfunction from systolic failure.11,13 A comparison of the left ventricular pressure-volume loop in systolic and diastolic dysfunction is shown in Figure 19-1. Systolic failure is characterized by higher than normal diastolic volume and low EF, whereas the pressure-volume loop in diastolic failure indicates poor compliance with a lower diastolic volume at a higher than normal pressure.
Compensatory Mechanisms and Remodeling
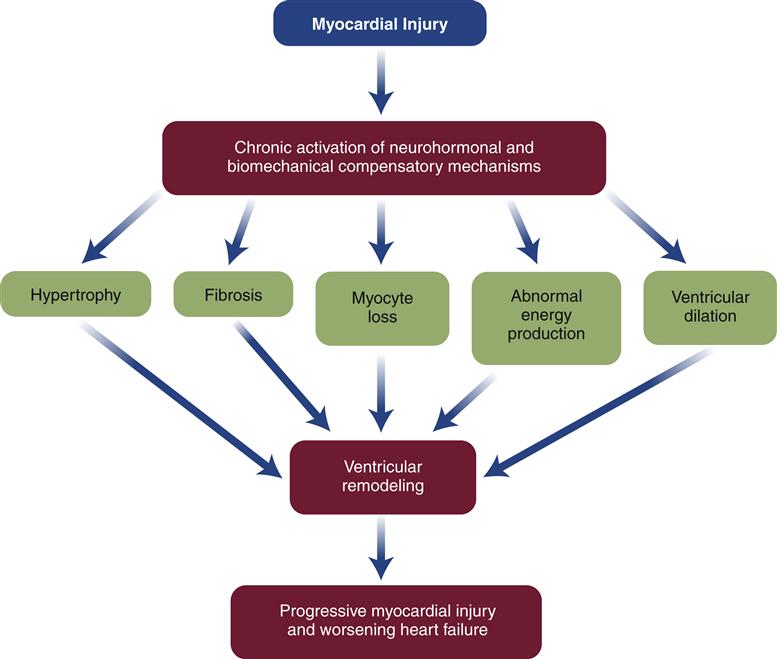
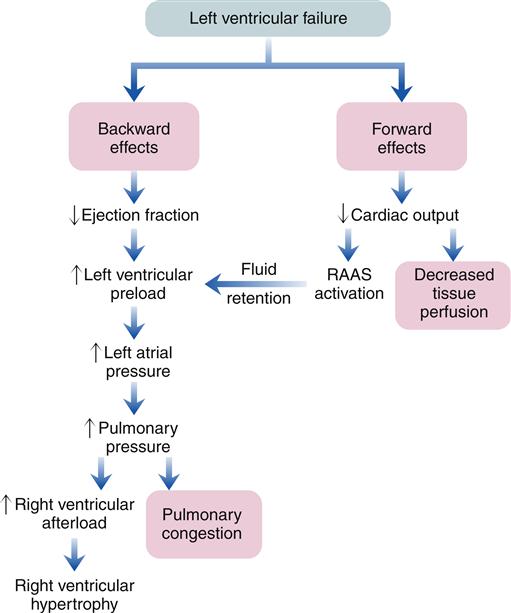
When the heart fails to provide adequate cardiac output to meet tissue demands, a number of compensatory mechanisms are triggered. In the short term, these mechanisms are helpful in restoring cardiac output toward normal levels, but in the long term, they are detrimental to cardiac structure and function. Much of the current management of HF is aimed at attenuating the harmful consequences of these compensatory responses.7 Three main compensatory mechanisms are activated in heart failure: SNS activation, increased preload, and myocardial hypertrophy (Figure 19-2).
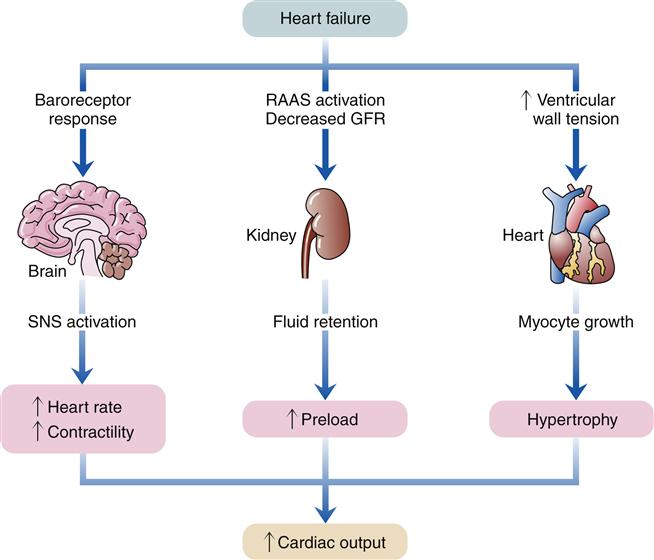
GFR, Glomerular filtration rate; RAAS, renin-angiotensin-aldosterone system; SNS, sympathetic nervous system.
Sympathetic Nervous System Activation
Sympathetic activation of the heart is primarily a result of baroreceptor reflex stimulation. The baroreceptors (pressoreceptors), located in the aorta and carotid arteries, detect a fall in pressure because of diminished stroke volume and transmit this information to the central nervous system (CNS). The CNS increases activity in the sympathetic nerves to the heart, resulting in increased heart rate and contractility. However, because of impaired contractile ability, the failing heart may have reduced responsiveness to sympathetic activation. Sympathetic activation also causes venoconstriction, which redistributes blood and increases cardiac preload. Sympathetic constriction of arterioles helps to maintain blood pressure when cardiac output is reduced. Specialized cells in the kidney called juxtaglomerular cells also receive SNS stimulation when cardiac output falls. The juxtaglomerular cells release renin and initiate the renin-angiotensin-aldosterone cascade, leading to salt and water retention by the kidney. Sympathetic activation is an early and immediate compensatory response to insufficient cardiac output.
Sympathetic activation is a very effective means for increasing cardiac output in an acute process, such as volume depletion. However, in heart failure, sympathetic activation becomes a chronic process that is ultimately deleterious. A major problem with excessive sympathetic activation is that afterload on the left ventricle can be increased significantly. A high afterload increases cardiac workload and may decrease stroke volume. Therefore, treatment of high blood pressure is important to improve cardiac function in patients with HF.8
Drugs that block β1 receptors have been advocated in the management of HF to inhibit the cardiac effects of sympathetic activation. Many clinicians had been reluctant to use β-blockers in patients with HF because these drugs are negative inotropes and have the potential to reduce cardiac output. In HF, where cardiac output is already low, the use of a negative inotrope would seem to be contraindicated. However, several randomized clinical trials have reported an improved mortality rate in patients receiving certain β1-blockers, and they are now recommended as standard therapy in most HF guidelines.6,13 Long-term SNS stimulation of the heart may contribute to heart failure progression and remodeling of the cardiac tissue. Remodeling is a process of myocyte loss, hypertrophy of remaining cells, and interstitial fibrosis (Figure 19-3). The remodeled tissue is less functional and may predispose to worsening failure and cardiac dysrhythmias.
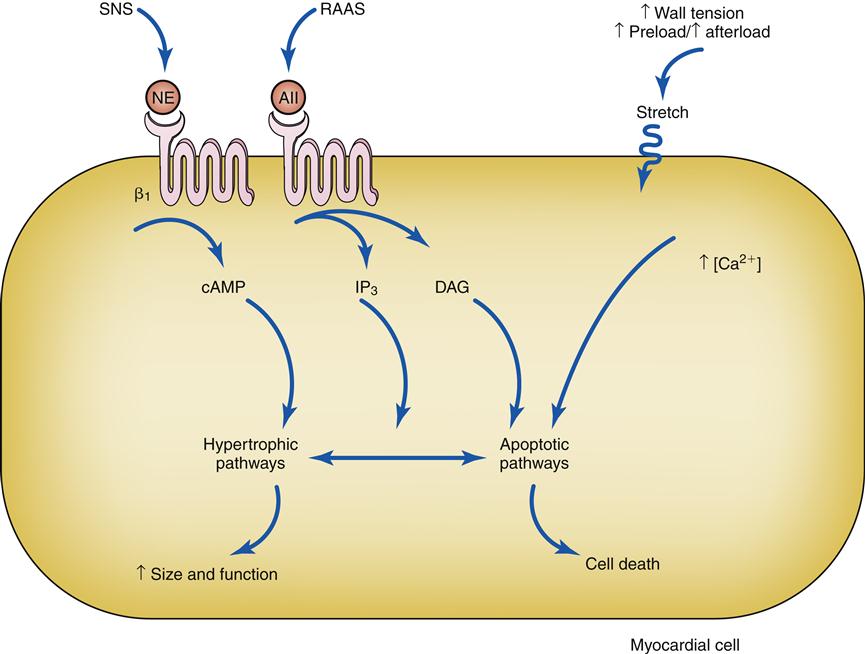
Activation of β1 receptors and AII receptors along with stretch of the cell membrane trigger signaling cascades. Under some conditions these triggers lead to effective hypertrophy and an increase in size and function, and in others they trigger apoptotic cell death. AII, Angiotensin II; cAMP, cyclic adenosine monophosphate; DAG, diacylglycerol; IP3, 1,4,5-inositol trisphosphate; NE, norepinephrine; RAAS, renin-angiotensin-aldosterone system; SNS, sympathetic nervous system.
Increased Preload
Increased preload in the cardiac chambers is initially a consequence of reduced EF with a resultant increase in residual end-systolic volume. Subsequently, decreased cardiac output to the kidney reduces glomerular filtration, resulting in fluid conservation. In addition, the renin-angiotensin-aldosterone system (RAAS) is activated because of reduced blood flow to the kidney and SNS activation of the juxtaglomerular cells. Angiotensin II (AII) and aldosterone enhance sodium and water reabsorption by the kidney, contributing to an elevated blood volume. Increased preload is a compensatory mechanism that enhances the ability of the myocardium to contract forcefully. An enlarged chamber volume causes the myocardial fibers to lengthen during diastole, which results in greater fiber shortening during contraction (Frank-Starling mechanism).12 The diastolic length of the muscle fibers is thought to determine the number of effective cross-bridge cycles that can be accomplished during systole.10 Thus, up to a point, an increase in the volume or preload of the heart will result in a greater force of contraction (Figure 19-4). The cardiac function curve flattens out at a certain point, and minimal benefit is obtained despite increasing preload. Patients with systolic failure have a cardiac function curve that is flat and shifted to the right of normal. Thus, they require a higher preload to achieve a given stroke volume. However, patients with HF often retain so much volume that their hearts are functioning on the flat part of the curve. These patients benefit from preload reduction, which will decrease systemic and pulmonary congestive symptoms and cardiac workload with little or no reduction in cardiac output. Diuretics are commonly used to achieve moderate preload reduction.
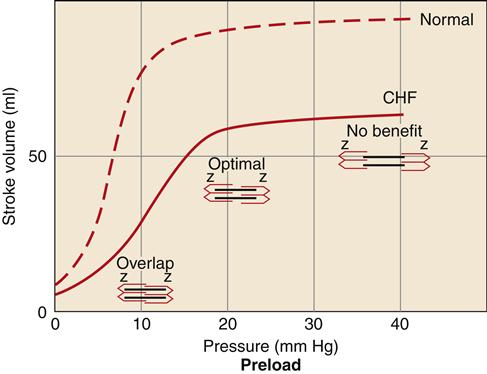
Systolic failure results in a shift of the curve to the right and a dampening of maximal stroke volume. A greater preload is required to achieve a given stroke volume compared with the normal ventricle. CHF, Congestive heart failure.
Myocardial Hypertrophy and Remodeling
Hypertrophy of cardiac muscle cells is the third mechanism of compensation and generally takes much longer to occur than preload enhancement or sympathetic activation. Hypertrophy appears to result, in part, from a chronic elevation of myocardial wall tension.10 Wall tension may be high as a result of increased diastolic blood volume (high preload) or as a consequence of high systolic pressures generated in the chamber (to overcome high afterload). The relationship between myocardial wall tension and intrachamber pressure and diameter is described by the law of Laplace: tension = (transmural pressure × radius)/wall thickness. When the ventricular chamber enlarges and pressures increase, more tension is created in the ventricular muscle wall (Figure 19-5). The development of high systolic pressures in the ventricle may be necessary to overcome a high afterload, such as occurs with arterial hypertension and aortic valve stenosis. The hypertrophy of contractile elements in the myocardium increases the heart’s pumping force and helps to reduce the wall tension of the heart toward normal levels. In general, an increase in chamber diameter because of excessive preload is thought to contribute to eccentric hypertrophy in which the muscle fibers elongate.7,10 High afterload results in concentric hypertrophy in which the muscle fibers grow in diameter and thicken the ventricular wall (Figure 19-6).
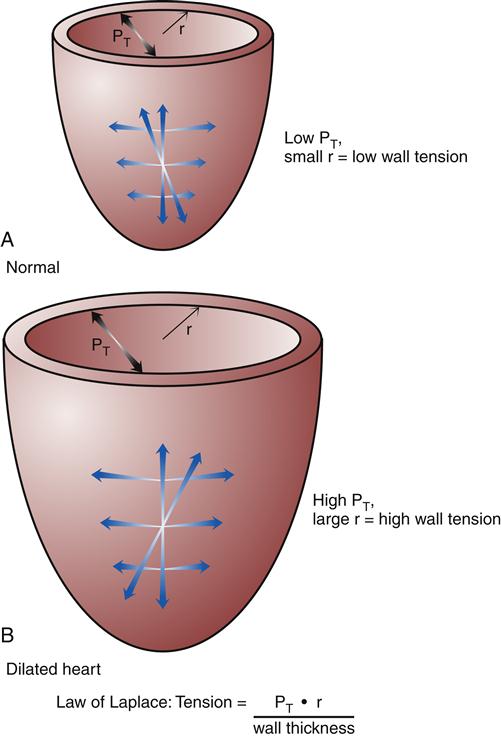
According to the law of Laplace, an increase in chamber radius or pressure will increase wall tension. The hypertrophic response increases wall thickness and helps to relieve wall tension. A, Heart with normal radius (r) and intraventricular pressure. B, Heart with enlarged chamber and high intraventricular pressure. PT, Transmural pressure.
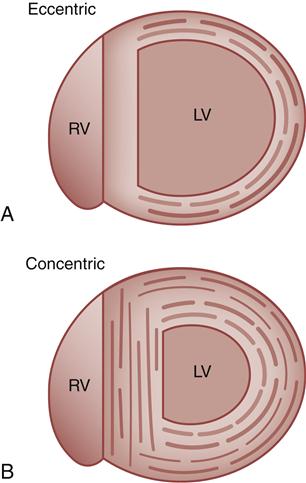
A, Eccentric, in which muscle fibers grow in length and the chamber diameter increases. B, Concentric, in which muscle fibers grow in diameter and the ventricular wall becomes thicker. LV, Left ventricle; RV, right ventricle.
Neurohormones, which include norepinephrine (NE) and angiotensin (Ang II), also have hypertrophic effects on the heart. Circulating Ang II levels are higher than normal in HF because of poor kidney perfusion, which triggers production of Ang II through the RAAS. In heart failure, Ang II is also produced locally in the heart. Ang II binds to the angiotensin type 1 (AT1) receptor on cardiac myocytes to activate genes in various growth pathways. Initially, hypertrophy may help the heart compensate for acute loss of myocardial tissue from MI or help maintain cardiac function during chronic hypertension. But over time, the signals that promote hypertrophy are thought to trigger a type of ventricular remodeling that contributes to progression of heart failure. Pathologic remodeling includes loss of myocardial cells through apoptosis and production of fibrous changes in the heart that stiffen the ventricles and contribute to diastolic failure (Figure 19-7). In addition to NE and Ang II, a number of immune cytokines have been implicated in cardiac remodeling.7,11,12
Evidence for a role of Ang II in remodeling comes from drug studies in which Ang II production is inhibited or the actions of Ang II are blocked at the AT1 receptor. An important enzyme in the pathway of Ang II production is angiotensin-converting enzyme (ACE). Drugs called ACE inhibitors (ACEIs) have been developed to inhibit the activity of this enzyme and prevent formation of Ang II. Another drug class, angiotensin type 1 receptor blockers (ARBs), binds to this Ang II receptor and blocks the intracellular actions of Ang II. ARBs were developed as a more selective means of RAAS inhibition, and to improve the safety and tolerability profile of ACE inhibitors. Both ACE inhibitors and ARBs have been shown to significantly reduce HF mortality, and both drug classes are used as standard therapy in HF.1,4,6 In general, ARBs are prescribed if a patient is intolerant of ACE inhibitors.
In summary, enhanced preload and cardiac hypertrophy may allow a heart to compensate for reduced ventricular function for an extended period. Unfortunately, these compensatory mechanisms, which serve to restore cardiac output to the tissues, also result in an increase in myocardial work and oxygen requirements and appear to cause pathologic remodeling. Progression and decompensation may occur when the primary disease plus the superimposed burdens of compensation overwhelm the heart’s ability to generate adequate contractile force. The focus of therapy for HF is to maintain a state of compensation by minimizing cardiac work while optimizing cardiac output and preventing or delaying ventricular remodeling.
Clinical Manifestations
The clinical presentation of HF differs depending on which ventricle (left, right, or both) is failing to pump blood adequately. Left ventricular failure is the most common presentation of HF. Because of circulatory dynamics, left ventricular failure often leads to right ventricular failure—a condition termed biventricular failure. The etiologic process, clinical manifestations, and management of isolated right ventricular failure differ substantially from those for left ventricular and biventricular failure. Recall that the right side of the heart receives blood from the systemic venous circulation and pumps blood into the pulmonary system, whereas the left side of the heart receives blood from the pulmonary circulation and delivers it to the systemic arterial system (Figure 19-8). Insufficient cardiac pumping is manifested by poor cardiac output, called forward failure, and by congestion of blood behind the pumping chamber, called backward failure. The clinical manifestations of left and right ventricular failure differ as a result of the anatomic location of the “backward” or congestive processes, but the forward effects of low cardiac output are the same.
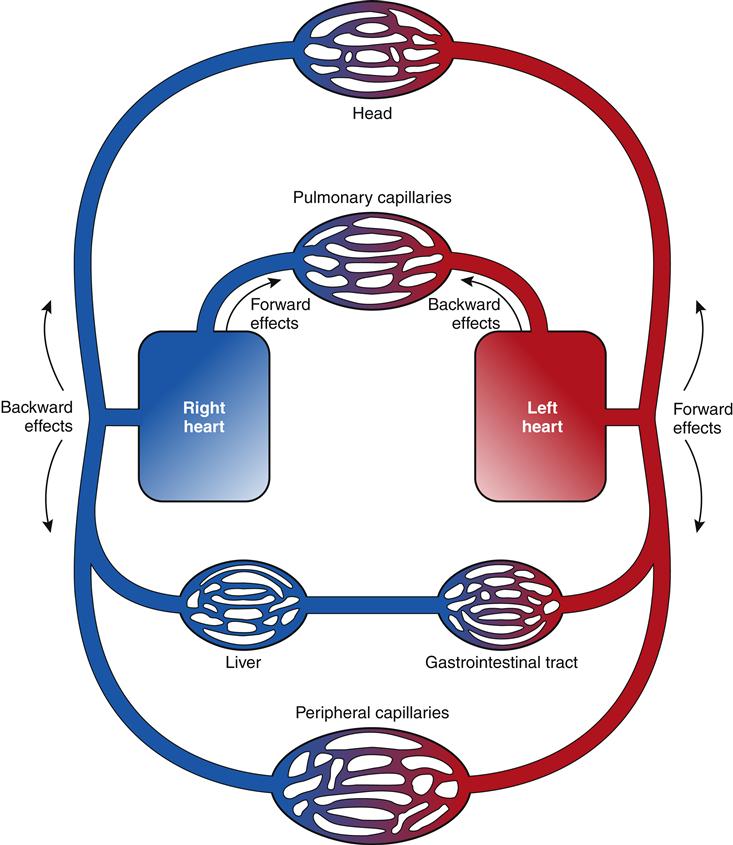
The forward effects of HF are due to insufficient cardiac output with diminished delivery of oxygen and nutrients to peripheral tissues and organs. Inadequate perfusion of the brain may lead to restlessness, mental fatigue, confusion, anxiety, and impaired memory. Generalized fatigue, activity intolerance, and lethargy may be present.
Reduced perfusion of the kidney results in a decline in urine output (oliguria) with subsequent fluid retention. Activation of the RAAS contributes to conservation of sodium and water by the kidney, and may also cause blood vessel constriction. Constriction of blood vessels serves to maintain blood pressure and redistribute reduced cardiac output to vital organs. However, this vasoconstriction also increases afterload, so the damaged left ventricle must generate more force to pump the same volume of blood. Depending on how much the afterload increases, the damaged left ventricle may not be able to pump sufficient quantities of blood into the circulation. If renal blood flow becomes severely limited, the patient with left ventricular failure may develop kidney failure. Forward failure also results in activation of the SNS because of the baroreceptor reflex. Sympathetic activation contributes to blood vessel constriction and helps maintain blood pressure in the face of reduced cardiac output; however, as with Ang II, SNS activation increases left ventricular afterload. SNS activation results in a compensatory increase in heart rate that may augment cardiac output to some extent, but also raises myocardial ATP consumption.
Left-Sided Heart Failure
Left-sided heart failure is most often associated with left ventricular infarction and systemic hypertension.7,8 The backward effects of left-sided heart failure may produce dramatic clinical symptoms attributable to pulmonary dysfunction (Figure 19-9). Ineffective pumping of the left ventricle results in an accumulation of blood within the pulmonary circulation. As hydrostatic pressure builds within the pulmonary veins and capillaries, fluid is forced from the capillaries into interstitial and alveolar spaces, causing edema. Pulmonary congestion and edema are associated with a number of clinical findings (Figure 19-10). Dyspnea, or breathlessness, occurs early in the progression of left-sided heart failure and may be considered the cardinal symptom. Difficulty breathing may be exacerbated by activity (dyspnea on exertion), lying down (orthopnea and paroxysmal nocturnal dyspnea), and blood volume expansion from excessive salt or fluid intake. Orthopnea and paroxysmal nocturnal dyspnea are due in part to a redistribution of blood volume from the periphery to the heart when the individual lies down. The failing left ventricle is unable to effectively pump extra volume, and pulmonary congestion is worsened. The severity of orthopnea may be quantified by the degree of head elevation (e.g., number of pillows) used to relieve dyspnea. Paroxysmal nocturnal dyspnea refers to intermittent attacks of severe dyspnea during the night and is a most distressing form of orthopnea. The individual experiences a feeling of suffocation and panic at not being able to overcome the dyspnea. Sitting or standing helps to relieve the dyspnea because blood pools in the extremities, reducing pulmonary hydrostatic pressure and congestion.
RAAS, Renin-angiotensin-aldosterone system.
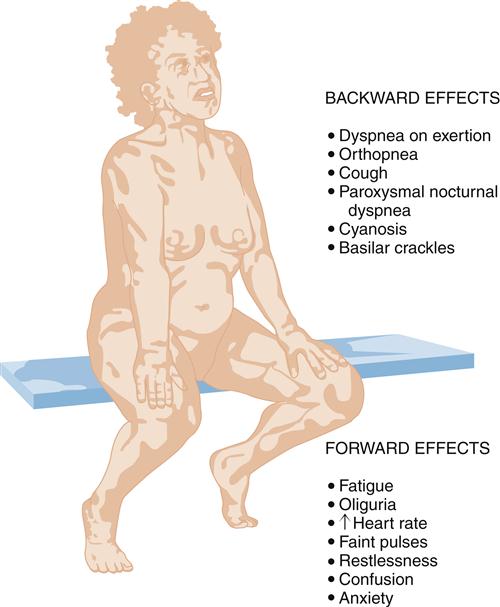
Clinical signs of pulmonary congestion include cough, respiratory crackles (rales), hypoxemia, and high left atrial pressure (LAP). Cough results from bronchial irritation associated with congestion. In severe cases, sputum may be blood tinged, from breakage of fragile capillaries, and frothy, from fluid buildup in the alveoli. The severity of pulmonary edema can be estimated from the location of crackles within the lung fields. Crackles are abnormal sounds caused by the movement of air through partially fluid-filled alveoli. Edema fluid collects in dependent lung fields because of gravity and progressively moves up the lung as more edema fluid accumulates. For example, in mild pulmonary edema, crackles might be heard with a stethoscope only at the base of the upright lung, but with increasing severity they become apparent in the lower third to half of the lung. Fluid in the alveoli and interstitial spaces also interferes with alveolar-capillary gas exchange and results in some degree of hypoxemia. Hypoxemia may be detected by arterial blood gas analysis or pulse oximetry and may be apparent clinically as cyanosis. Cyanosis refers to a blue coloration of the skin typically seen around the mouth (circumoral cyanosis) and results from the presence of significant amounts of desaturated hemoglobin in the blood. Cyanosis is a late sign, and is clinically evident only when a large amount (about 5 g/dl) of hemoglobin is deoxygenated (less than or equal to 75% saturated).
Elevated LAP is a common finding in left-sided heart failure because of excessive blood volume and the compensatory responses of atrial dilation and hypertrophy. Atrial pressure can be estimated by inserting a balloon-tipped catheter (Swan-Ganz) into the pulmonary artery. If LAP acutely increases to 25 mm Hg (normal is 4 to 12 mm Hg), increased capillary filtration leads to pulmonary edema. Patients with chronic elevations in LAP associated with chronic HF are more resistant to developing acute pulmonary edema, and may not experience symptoms until pressures approach 40 mm Hg.7 On x-ray, findings of fluid overload include an enlarged heart and engorged pulmonary capillaries and lymphatic vessels.
Acute cardiogenic pulmonary edema is a life-threatening condition associated with left ventricular failure that severely impairs gas exchange, producing dramatic signs and symptoms. The patient exhibits severe dyspnea and anxiety, and a bolt-upright posture is usually assumed in order to maximize respiratory effort. Bubbly crackles may be heard all the way up the lung from the bases to the apices, and pink frothy sputum may be expectorated or well up from the trachea into the nose and mouth. Anxiety and hypoxemia contribute to tachycardia, which may worsen the pumping efficiency of the failing heart. Cyanosis and symptoms of tissue hypoxia are usually apparent. The immediate treatment is aimed at reducing the fluid volume in the lungs and supporting oxygenation.
Right-Sided Heart Failure
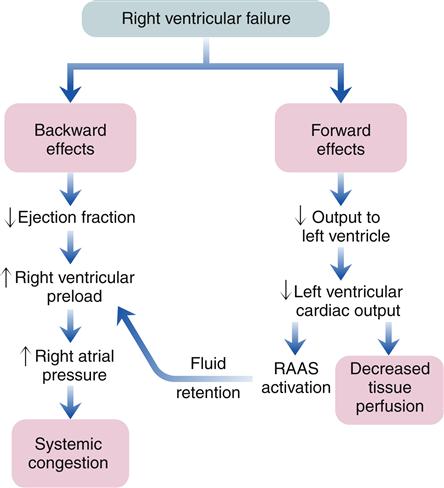
Because the right and left ventricles function in series, left ventricular failure eventually increases the workload on the right ventricle. Consequently, the right ventricle may fail. The etiology of right ventricular failure must include all the causes of left ventricular failure. Isolated right ventricular failure is rare and is usually a consequence of right ventricular infarction or pulmonary disease. Only 3% of MIs occur in the right ventricle; however, right ventricular infarctions are often poorly tolerated and difficult to manage.7 Pulmonary disorders that result in increased pulmonary vascular resistance impose a high afterload on the right ventricle. The resultant right ventricular hypertrophy, called cor pulmonale, may progress to right ventricular failure as the lung disease worsens.
Any lung disorder that decreases the total cross-sectional area of the lung vasculature can increase pulmonary vascular resistance and produce right ventricular strain. Hypoxemia, for example, causes the pulmonary arterioles to constrict, which increases pulmonary resistance. Constriction or blockage of the vascular bed, such as occurs with pulmonary hypertension or pulmonary embolus, similarly reduces the cross-sectional area of the pulmonary vasculature and leads to increased pulmonary resistance. If the increase in pulmonary resistance and right ventricular workload occurs gradually, the right ventricle can compensate by increasing preload and hypertrophy. However, the thin musculature of the right ventricle has limited ability to adjust to acute changes in workload, as would occur with a right ventricular infarction or large pulmonary embolus.
As with left-sided heart failure, congestion of blood occurs behind the failing right ventricle because of inefficiency of the pump. The backward effects of right-sided heart failure are due to congestion in the systemic venous system (Figure 19-11). Systemic venous congestion results in impaired function of the liver, portal system, spleen, kidneys, peripheral subcutaneous tissues, and brain (Figure 19-12).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


