- Vancomycin is used for the treatment of meticillin-resistant Staphylococcus aureus (MRSA) infections.
- The glycopeptides bind to D-ala-D-ala and form a ‘protective layer’, thus preventing some of the steps involved in cell wall assembly.
- Resistance to vancomycin can involve the production of an alternative ligase, which produces D-ala- D-lac. D-ala-D-lac performs the same role in cell wall assembly as D-ala-D-ala but has greatly reduced binding affinity for the glycopeptides.
The glycopeptide antibiotics, teicoplanin (a mixture of nine components: A2-1–A2-5 and RS-1–RS-4) and vancomycin (Figure 5.2.1), are used in the prophylaxis and treatment of severe infections caused by Gram positive bacteria which are penicillin-resistant (or in individuals who are penicillin-intolerant). The therapeutic indications for these antibiotics are given in Table 5.2.1.
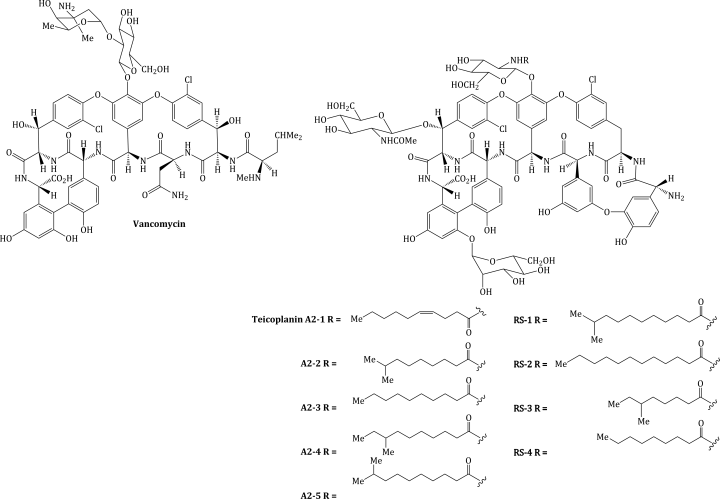
Table 5.2.1 Therapeutic indications for the glycopeptide antibiotics.
| Glycopeptide antibiotic | Indications |
| Teicoplanin | Serious Gram positive infections, e.g. septic arthritis or MRSA |
| Vancomycin | Endocarditis MRSA, Clostridium difficile-associated diarrhoea |
5.2.1 Discovery
Vancomycin was discovered when, in 1952, a missionary in Borneo sent a soil sample to a friend at the pharmaceutical giant Eli Lilly and Co. It was soon found that Streptomyces orientalis (also referred to as Amycolatopsis orientalis), an organism present in the sample, produced a compound which had activity against most Gram positive (including penicillin-resistant staphylococci), and some anaerobic, organisms. Originally referred to as 05865, this compound is now known as vancomycin, with its name being derived from the word ‘vanquish’ (a strong claim, which could easily have been the kiss of death for the drug) (Levine, 2006).
Like vancomycin, the teicoplanins were discovered from a soil isolate, this time from an organism called Actinoplanes teichomyceticus (Parenti et al., 1978).
5.2.2 Synthesis
The structural complexity of these antibiotics, with their cyclic rings incorporating unusual amino acids, unnatural sugar units, atropisomerism,17 and numerous stereogenic centres (nine in the vancomycin aglycone18 and eight in that of teicoplanin), meant that the elucidation of their structure was a slow process (Barna and Williams, 1984). This structural complexity also means that total synthesis of these antibiotics, while fascinating from an academic viewpoint, will, once again, never be able to produce sufficient quantities for clinical use. The challenge associated with the total synthesis of the glycopeptides antibiotics has again attracted some of the world’s leading synthetic chemists, and the first reported synthesis of the aglycone portion, with the longest linear synthetic sequence consisting of 40 steps, is outlined in Scheme 5.2.1 (Evans et al., 1998).
Scheme 5.2.1 Building blocks used in the Evans synthesis of the vancomycin aglycone (Evans et al., 1998)
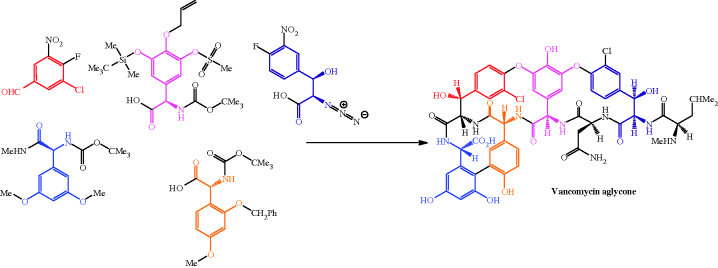
The availability of the vancomycin aglycone soon led to the total synthesis of vancomycin and the possibility of the incorporation of alternative disaccharide units (Thompson et al., 1999). As mentioned above, these total syntheses were never going to be sufficient for the production of sufficient quantities of the drug, and vancomycin is produced on an industrial scale by the fermentation of A. orientalis (Padma et al., 2002).
Teicoplanin has also been prepared by a total synthesis (Boger et al., 2000), but is produced on an industrial scale by a submerged culture of A. teichomyceticus. Rather unusually, this microorganism is sensitive to the antibiotic it produces (a bit of a design fault, to say the least) (Jung et al., 2009) and so innovative methods have been employed to enable large-scale production, including mutagenesis so that the bacterial cells become more tolerant to teicoplanin (Jung et al., 2008) and, once it is formed, trapping the antibiotic on an absorbent resin (Lee et al., 2003).
5.2.3 Bioavailability
As a result of its multiple anionic and cationic charged groups and hydrophilicity, vancomycin is poorly absorbed from the gastrointestinal (GI) tract and has a bioavailability of less than 2% (Anderson et al., 2001). In addition, the plasma half-life of vancomycin is 3–13 hours (Felmingham, 1993), so it is usually administered IV, with doses of 500 mg every 6 hours (or 1 g every 12 hours). For twice-daily dosing, the trough level should be 15–20 mg/L as this minimises the chances of resistance developing (Rybak et al., 2009). No relationship appears to exist between the peak concentrations, trough concentrations, and pharmacodynamic parameters (such as time above the MIC or AUC/MIC) and the eradication of the microorganism eradication or patient outcome (Rybak, 2006). Oral vancomycin is used only for the treatment of Clostridium difficile-associated diarrhoea as, being poorly absorbed, the orally administered vancomycin remains in the gut and so acts very effectively on the C. difficile present.
Vancomycin is eliminated by the kidneys (with 80–90% recovered unchanged in urine within 24 hours of a single dose), so the dose should be altered in renally impaired patients, using the patient’s creatinine clearance rate to determine the appropriate dose and schedule.
Vancomycin penetrates into most body spaces, with values for the apparent volume of distribution of 0.4–1 L/Kg (Rybak et al., 2009).
Teicoplanin is also excreted by the kidney but is 90% bound to serum albumin (protein binding for vancomycin is less than 50%) and, as it has a significantly longer half-life, only requires once-daily administration (Felmingham, 1993; Yu et al., 1995).
For the same reasons of ionisation, hydrophilic nature, and large size that confer poor oral bioavailability, vancomycin and teicoplanin do not traverse the Gram negative bacterial membrane, which is presumably why they are inactive against these species. As their site of action is the bacterial cell wall (as explained in Subsection 5.2.4) and the cell wall of Gram positive bacteria is less complex and so easier to breach, the glycopeptide antibiotics are able to access their site of action sufficiently well to exert their bacteriostatic effect. For more on the bioavailability of the glycopeptides, see Rybak et al. (2009).
5.2.4 Mode of Action and Selectivity
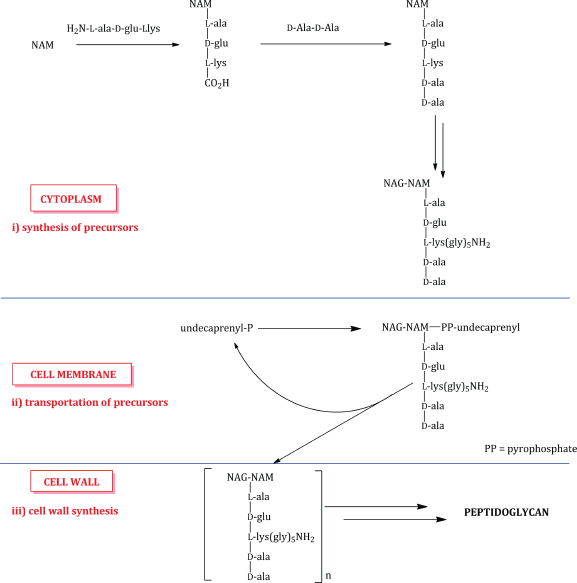
The glycopeptides interfere with cell-wall peptidoglycan synthesis as a result of their binding affinity for the D-ala-D-ala amino acid sequence present in the transpeptidation precursors. As you will remember from the introductory section, the biosynthesis of peptidoglycan involves three main stages: i) synthesis of the cell-wall precursors, ii) transportation of the precursors across the cell membrane by a carrier lipid (undecaprenyl in this case), and iii) cell-wall synthesis, involving coupling of the precursors into polymeric chains and the crosslinking of these chains (Scheme 5.2.2). As eukaryotic cells do not have a cell wall, the glycopeptides are selective bactericidal agents.
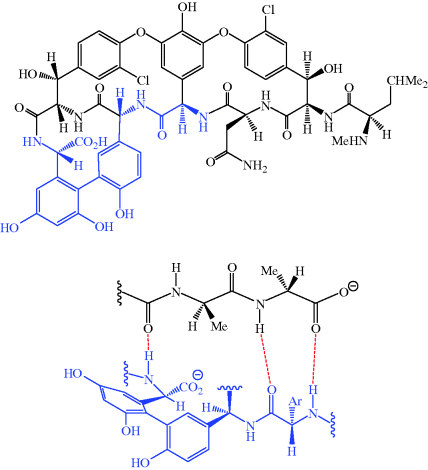
The glycopeptides contain hydrogen-bond donor and acceptor groups and the aglycone portion forms five hydrogen bonds to the terminal D-ala-D-ala sequence of the precursor pentapeptide chain (three of these hydrogen bonds are shown in Figure 5.2.2) (Barna and Williams, 1984).
Figure 5.2.2 Some of the hydrogen bonds (red) formed between vancomycin and D-ala-D-ala. The portion of vancomycin corresponds to the region highlighted in blue in the full structure of the aglycone
By binding to the D-ala-D-ala sequence, the glycopeptides inhibit a number of processes which are involved in the assembly of the cell wall, but they do not do this by directly acting on the enzymes responsible; instead they form a ‘protective layer’ preventing these steps, with the D-ala-D-ala unit being held within a deep cleft of the antibiotic. One of the earliest steps in cell-wall synthesis involves the transglycosylase-catalysed transfer of a disaccharide peptide from its lipid carrier to the growing peptidoglycan chain. Despite being bound to a site of this disaccharide-peptide unit that is remote from the disaccharide portion, these antibiotics inhibit this process, possibly through a steric clash with some region of the transglycosylase enzyme lying close in space to the terminal D-ala-D-ala of the peptide chain (Scheme 5.2.3a) (Reynolds, 1989).
Scheme 5.2.3 Schematic representation of vancomycin inhibition of (a) transglycosylase and (b) peptidoglycan transpeptidase activity
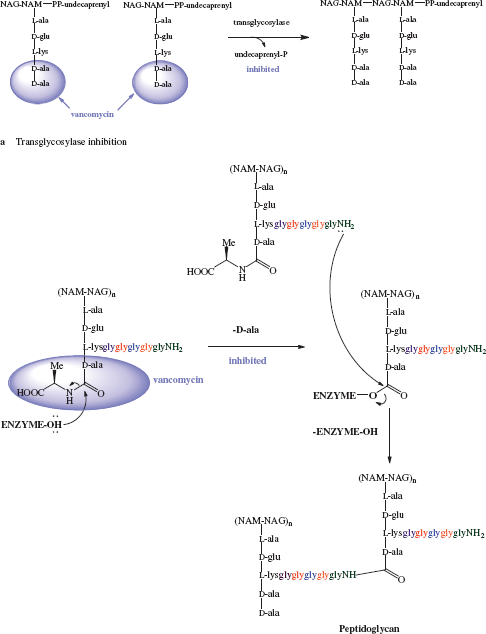
A later step in the formation of the cell wall is the penicillin-binding protein-catalysed removal of the terminal D-ala residue (by D-alanine carboxypeptidase). This process involves the formation of an acyl-enzyme intermediate, which is subsequently attacked by a glycine amino group to give a new peptide bond, thus crosslinking the peptidoglycan chains (catalysed by peptidoglycan transpeptidase, also called PBP) (Scheme 5.2.3b). As the glycopeptide antibiotics bind to the D-ala-D-ala sequence, they once again prevent this process from taking place (Reynolds, 1989).
In summary, as a result of vancomycin’s binding to the D-ala-D-ala sequence at the end of the pentapeptide chain of the peptidoglycan precursor, transglycosylase and transpeptidase activity are inhibited and cell-wall formation is disrupted.
5.2.5 Bacterial Resistance
Now that we know that the glycopeptide antibiotics act by specifically binding to the D-ala-D-ala dipeptide sequence, it should come as no surprise that bacterial resistance to these agents involves the modification of this sequence through alterations in the ligase activity (the enzyme which joins the two amino acids to produce the dipeptide).
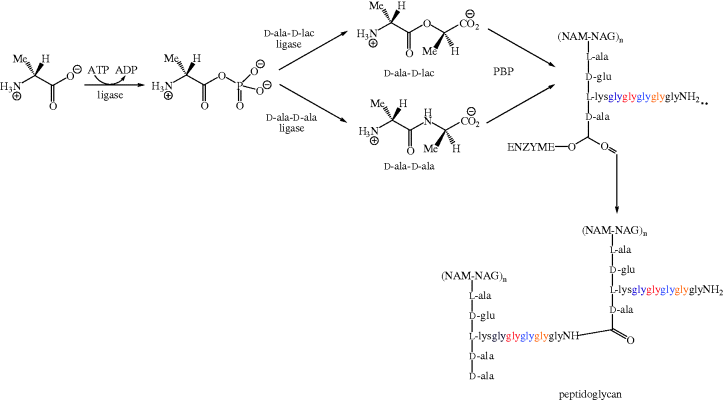
There are five types of glycopeptide resistance in vancomycin-resistant enterococci (VRE), classified as VanA, B, C, D, and E. The first four of these are fairly well understood, and share some degree of similarity, so we will concentrate on the VanA phenotype, in which resistance is mediated by the transposon containing the nine genes of the vanA gene cluster. Four of these genes are responsible for the molecular processes giving rise to vancomycin resistance; vanH produces VanH, a dehydrogenase which reduces pyruvate to D-lactate (D-lac), for subsequent incorporation into D-ala-D-lac by VanA (the product of the vanA gene), a D-ala-D-lactate ligase which synthesises an ester (D-Ala-D-lac) rather than an amide (D-ala-D-ala) (Table 5.2.2). The D-ala-D-lactate sequence replaces D-ala-D-ala in the pentapeptide chain (the resulting chain is now not, of course, a pentapeptide). Crucially, D-ala-D-lac has 1000-fold reduction in affinity for vancomycin (being able to form fewer hydrogen bonds) but can still be attacked by glyNH2 and so act as precursor for crosslink formation (Scheme 5.2.4) (Malathum and Murray, 1999). Such altered ligases are also produced by vancomycin-producing microorganisms (Kuzin et al., 2000).
Table 5.2.2 VRE VanA resistance phenotype genes and their enzyme product/function.
| Gene | Enzyme product and function |
| vanS | VanS (membrane-associated sensor kinase,a which recognises the presence, or an effect, of vancomycin) |
| vanR | VanR (cytoplasmic response regulator which receives a signal from VanS then activates its own promoter and those of VanH, VanA, and VanX) |
| vanH | VanH (dehydrogenase responsible for reduction of pyruvate to D-lactate) |
| vanA | VanA (D-ala-D-lac ligase which utilises the D-lactate product of VanH) |
| vanX | VanX (a D,D-dipeptidase which hydrolyses D-ala-D-ala to individual D-ala units, thus reducing the cellular quantities of the dipeptide unit and preventing its incorporation into the pentapeptide chain) |
| vanY | VanY (a D-carboxypeptidase which removes the terminal D-ala from the pentapeptide chain to give a tetrapeptide) |
| vanZ | Function unknown, confers low-level resistance to teicoplanin only |
| ORF1 | Transposase which binds to the end of transposon Tn1546 and catalyses its movement to another part of the genome |
| ORF2 | Resolvase which is responsible for the recombination of transposon Tn1546 and the bacterial genome |
| a Kinases catalyse the phosphorylation of a substrate. | |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


