Genomics and the Kidney
LEARNING OBJECTIVES
INTRODUCTION
Chronic kidney disease (CKD) afflicts at least 21 million persons in the United States.1 The majority of CKD is due to longstanding and uncontrolled diabetes mellitus and hypertension. In addition to treatments aimed at reducing blood sugar and blood pressure, other common approaches for preserving kidney function include angiotensin-converting enzyme inhibitors (ACEIs) and angiotensin receptor blockers (ARBs) for reducing proteinuria, immunosuppressants and cytotoxic compounds for glomerulonephritis, and immunosuppressants for kidney transplantation. There has been a large expansion of studies within the nephrology research community to identify genetic variants that pose a risk for a particular form of CKD or serve as a risk factor for its progression (Table 17B–1). The role of genetic polymorphisms on drug metabolism and disposition (DMD) in patients with CKD has more recently gathered interest. This aspect is important as CKD, independent of genetic variants, is known to influence both the renal and non-renal clearance of drugs. Together, genomic and nongenomic factors could considerably influence the pharmacokinetics of drugs and overall responses to treatments in the CKD population. This chapter undergoes an extensive review of genomics in order that a comprehensive understanding of its role in nephrology is provided to clinicians and researchers (Figure 17B–1). This information will be necessary for clinicians as the field evolves to treatments targeted toward genes or aberrant proteins. Additionally, clinicians will need to understand how to provide adequate therapy regimens in the face of physiological and genetic alterations that are present in CKD patients.
TABLE 17B–1 Chronic kidney diseases with associated genetic polymorphisms.
FIGURE 17B–1 Implications for genomics and kidney diseases. DMD, drug metabolism and disposition.
DRUG-INDUCED NEPHROTOXICITY
Data are emerging that implicate a role for genetic polymorphisms in drug-induced nephrotoxicity. This is important since nephrotoxicity due to drugs contributes to between 8% and 60% of acute kidney injury (AKI) cases.2 In adult and pediatric intensive care units, prescriptions for nephrotoxic drugs account for ∼25% of all drug orders.3 Kidney biopsies are not standard of care to diagnose suspected AKI due to drugs. However, the diagnosis of nephrotoxicity is usually classified according to the kidney location or process affected by the toxicity, for example, hemodynamic, glomerular, tubular, tubulointerstitial, and obstructive. Clinically, nephrotoxicity is manifest as a rise in serum creatinine and blood urea nitrogen several days after exposure to drugs that cause the initial insult to the kidney. Newer, sensitive markers such as kidney injury molecule 1 (Kim-1), neutrophil gelatinase–associated lipocalin (NGAL), N-acetyl-β-glucosaminidase (NAG), clusterin, cystatin C, and β2 microglobulin can detect nephrotoxicity earlier in its clinical course, with suggestions that these improvements could enable damage to be detected within several hours after an initial insult. This is advantageous to the patient, as offending drugs can be discontinued early and/or strategies, such as appropriate hydration, and/or dosage modification can be implemented in an expeditious manner.
Clinicians are seeking early detection and improved prediction tools that may be useful in eliminating or reducing the impact of drug-induced nephrotoxicity. Pharmacogenomics and toxicogenomics are emerging areas that have gained interest in nephrotoxicity due to drugs and chemicals. The possibility of preemptively screening patients for polymorphisms that may enhance the risks of nephrotoxicity on exposure to a particular drug, prior to therapy initiation, is appealing. While this area of research is currently evolving, there are several publications that support a role for genetic polymorphisms in drug metabolism and/or drug transport pathways as risk factors for nephrotoxicity. The kidney itself is a high-risk candidate for toxicities and drug interactions secondary to genetic polymorphisms due to the presence of numerous transporters and metabolizing enzymes in this eliminating organ (Figures 17B–2 and 17B–3). In general, for transport proteins localized to the nephron, polymorphisms that increase the activity of efflux transporters and decrease the activity of uptake transporters are protective for nephrotoxicity. The nephrotoxicity risk of enhanced-activity versus reduced-activity polymorphisms in drug metabolism genes is dependent on whether the parent or a metabolite is implicated as the main nephrotoxic component. Other genes, in addition to those regulating drug metabolism and transport, have also been suggested to contribute to nephrotoxicity. This section will describe the available information concerning the role of genomics in nephrotoxicity due to immunosuppressants of the calcineurin inhibitor class, for example, cyclosporine and tacrolimus, and the chemotherapy agent cisplatin.
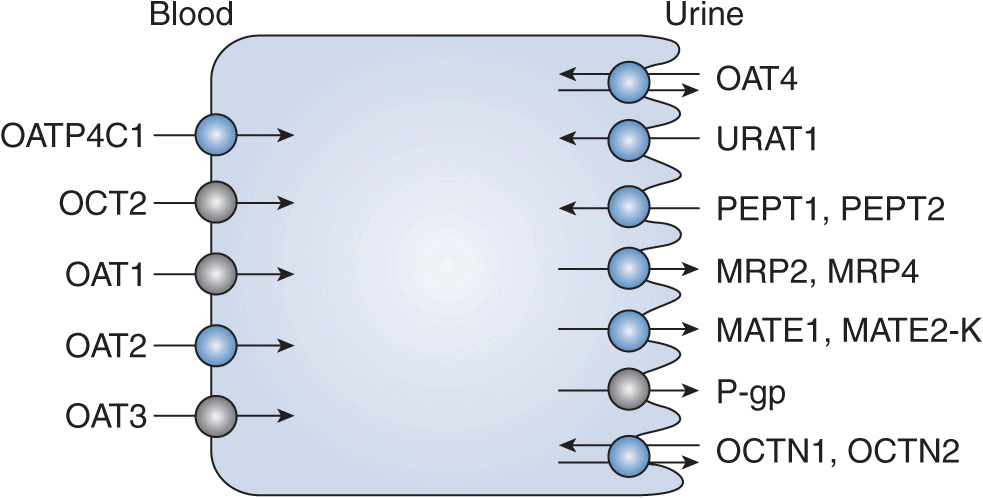
FIGURE 17B–2 Proximal tubule and transporters: kidney locations for pharmacogenomic effects. MATE, multidrug and toxin extrusion transporter; MRP2, multidrug resistance-associated protein transporter; OAT, organic anion transporter; OATP, organic anion transporting polypeptide; OCT, organic cation transporter; OCTN, carnitine/organic cation transporter; PEPT, peptide transporter; P-gp, multidrug resistance transporter; URAT, urate transporter.
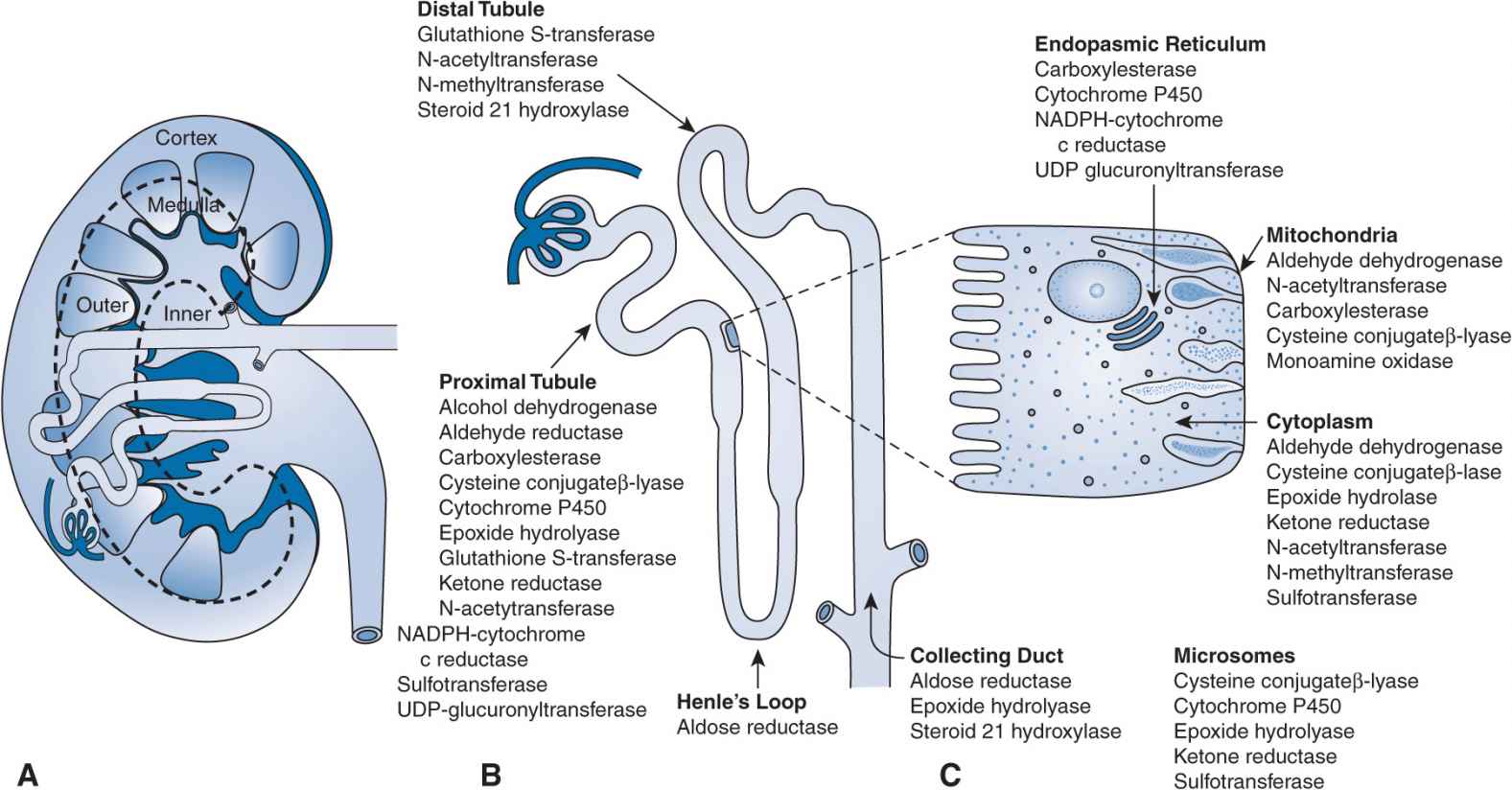
FIGURE 17B–3 (A–C) Drug metabolism in the kidney: locations for pharmacogenomic effects. NADPH, nicotinamide adenine dinucleotide phosphate; UDP, uridine diphosphate.
Calcineurin Inhibitors
Calcineurin inhibitors are used therapeutically in transplant recipients and patients with autoimmune diseases. Nephrotoxicity due to calcineurin inhibitors can be acute, represented by thrombotic microangiopathy and tubular vacuolization, or chronic, represented by interstitial fibrosis, glomerulosclerosis, and tubular atrophy. It is suggested that 20–94% of solid organ transplant patients who are prescribed calcineurin inhibitors will develop nephrotoxicity.4–6 Clinical risk factors for nephrotoxicity secondary to calcineurin inhibitors have included the exposure to the kidney tubules as mediated through transporters, for example, P-glycoprotein, and through drug metabolism enzymes, for example, CYP3A5. Due to some data linking alterations in exposure to cyclosporine or tacrolimus and presence of certain genetic polymorphisms in the genes encoding P-glycoprotein (ABCB1) and CYP3A5 (CYP3A5),7–11 it is reasonable to ascertain that polymorphisms may contribute to the risks of nephrotoxicity secondary to influencing localized drug exposure. In fact, local kidney exposure to cyclosporine has a direct relationship with increased histologic damage as well as decreased kidney function.12 Additionally, altered kidney expression of P-glycoprotein and CYP3A5 proteins predispose transplant patients to histologically confirmed nephrotoxicity.13,14
Human studies have demonstrated susceptibility to calcineurin inhibitor nephrotoxicity based on CYP3A5 and/or ABCB1 genotype.15–20 For tacrolimus, liver donor CYP3A5*3/*3 genotype (A6986G, rs28383472) was associated with kidney dysfunction.15 However, the CYP3A5*1 allele was associated with nephrotoxicity in recipients of kidney allografts.18 Kidney transplant recipients receiving tacrolimus who exhibited CYP3A4*1/CYP3A5*1 or CYP3A4*1B/CYP3A5*1 haplo-types had a higher risk of biopsy-proven nephrotoxicity versus those who exhibited the CYP3A4*1/CYP3A5*3 haplotype (for CYP3A4*1; A-392G, rs2740574).17 Liver transplant patients with variants in ABCB1 (G2677T, C3435T, and C1236T) had a higher incidence of kidney dysfunction due to tacrolimus.20 Hebert et al. supported these findings, reporting an increased frequency of kidney dysfunction in liver transplants patients receiving calcineurin inhibitors who had the ABCB1 2677TT genotype.21 While kidney transplant donor ABCB1 3435TT genotype was also found to be a risk factor for cyclosporine nephrotoxicity,19 ABCB1 genotype has not been linked with nephrotoxicity in heart transplant patients receiving cyclosporine.22
The existing studies also suggest that numerous nongenetic factors (transplant type, donor vs. recipient organ of metabolism vs. toxicity, administered drug, etc.) may account for the variable influences of CYP3A5 and ABCB1 genotypes on nephrotoxicity due to calcineurin inhibitors. In addition to DMD genes, polymorphisms in several other genes have been associated with calcineurin inhibitor nephrotoxicity.23–27
Cisplatin
Cisplatin is used in the treatment of various cancers of solid organs including head/neck, lung, and cervix. Representative oncology regimens commonly include cisplatin administration every 14-28 days. Nephrotoxicity is common, with damage to the kidney tubules occurring in 30-38% of patients after a single dose of cisplatin.28–30 Several preclinical studies suggest altered expression of OCT1/2, MRP2, and MATE1/2 transporters.31–33 SLC22A2 encodes the OAT2 protein, which is an uptake transporter of cisplatin located on the basolateral membrane of kidney proximal tubule cells. The G808T polymorphism (rs316019) in SLC22A2 is associated with reduced toxicity to the kidneys.32 These data suggest the role of genetic polymorphisms in drug transporter genes in modulating cisplatin nephrotoxicity.
Cisplatin is metabolized by several intracellular glutathione S-transferases (GSTs) to glutathione derivatives. Variants in GSTA1 C-69T (rs3957356) and GSTP1 A313G (rs1695) are associated with survival of cervical cancer patients34 and GSTP1 A313G and ABCC2 C-24T correlate with enhanced efficacy in patients with non-small cell lung cancer35,36 receiving cisplatin therapy. While these former GST polymorphisms have not been associated directly with cisplatin nephrotoxicity, it is plausible given that cellular toxicity from drugs often occurs in treatments that result in enhanced efficacy.
Genes involved in immunology, inflammation, and apoptosis have also been implicated in cisplatin nephrotoxicity. A genetic study in humans sought to evaluate SNPs that had demonstrated an enhanced susceptibility of cells to cisplatin.37 These evaluations reported genetic variants in cell recognition and adhesion (CDH13), metal-ion binding (ZNF659), transcription factor activity (LRRC3B, PITX2), and RNA binding (LARP2) with cisplatin sensitivity.37 While the available genomic studies suggest a link between polymorphisms in DMD and non-DMD genes and nephrotoxicity, prospective studies conducted in humans receiving cisplatin therapy are needed to more fully evaluate which polymorphisms are risk factors for the occurrence of clinical and subclinical nephrotoxicity.
ALBUMINURIA AND END-STAGE KIDNEY DISEASE
The National Kidney Foundation guidelines (K/DOQI) define the staging of CKD based on the presence of functional and/or structural abnormalities in the kidney.38 While functional variations are based on clearance represented by calculations employing serum creatinine,39,40 structural abnormalities are represented by the concentrations or amounts of urinary proteins, especially albumin, that are excreted into the urine.38 Albumin excretion has also been proposed as a biomarker for AKI.41 While albumin loss in the urine has classically been viewed as a marker for a problem in the filtration barrier at the level of the glomerulus, modern views also recognize the relevance of the proximal tubule and its protein reabsorptive function in proposing the use of albumin as a marker of tubule function as well. Proteinuria is viewed as a prognostic variable for the progression of kidney disease,42 a predictor of cardiovascular disease,43 and a predictor for all-cause mortality.44 Several studies have linked inflammation to albuminuria.45–47 Research related to genes that may be involved in the appearance of albuminuria has primarily focused on inflammation pathways, cubilin (CUBN), and nonmuscle myosin heavy chain 9 (MYH9). Other studies have been conducted to evaluate the role of polymorphisms within the genes of the renin–angiotensin–aldosterone system (RAAS) or in DMD pathways, for prediction of responsiveness to therapies employing ACEIs or ARBs in the reduction of urinary albumin (and protein) excretion.
A recent publication has focused on the contribution of polymorphisms in inflammatory response genes to albumin-uria.48 DNA was collected from 5,321 participants in the Third National Health and Nutrition Examination Survey (NHANES III). The associations between urinary albumin excretion and genetic variants were analyzed. The full models fit to the data incorporated adjustments for age, sex, alcohol consumption, educational attainments, and waist to hip ratio. Genetic variants that were important in predicting urinary albumin excretion were different according to race, defined as non-Hispanic whites, non-Hispanic blacks, and Mexican Americans. In non-Hispanic whites, full models demonstrated that variants in immunoglobulin Fc receptor (FCGR2A His166Arg; His167Arg, rs1801274), fibrinogen (FBG A-462G, rs1800790), mannose-binding lectin 2 (MBL2 G-618C; G-550C, rs11003125), nitric oxide synthase (NOS2A T-2892C; T-1173C, rs9282799), tumor necrosis factor (TNF A-555G, rs1800750 and A-417G; A-238G, rs361525), and glucokinase regulator (GCKR Pro446Leu, rs1260326) genes resulted in threshold P-values <.05. In non-Hispanic blacks, threshold P-values were shown in models for variants in c-reactive protein (CRP 3′UTR A/G, rs1205; downstream G/A, rs2808630; promoter T/A, rs3093058), thrombin (F2 upstream A/G, rs1799963), vitamin D receptor (VDR, Ile352Ile, rs731236), and transforming growth factor beta-1 (TGFB1 A-800G, rs1800468) genes. Lastly, for Mexican Americans, variants in NOS2A (promoter C/G, rs1800482), NOS3 (Asp298Glu, rs1799983; and C-786T, rs2070744), CRP (upstream G/A, rs11265260; downstream A/C, rs3093075), interleukin 1-beta (IL1B C-2022G, rs1143623), and TNF (A-487G; A-308G, rs1800629) genes resulted in models demonstrating P-values <.05. Further evaluations are needed to discern the potential mechanistic roles for the identified genes in contributing to an albuminuria phenotype. These results suggest that protein products of targeted genes involved in the inflammation process contribute to albuminuria through various physiological processes.
Cubilin
A missense variant (I2984V, rs1801239) of the CUBN gene, representing the protein cubilin, has recently been identified as a gene locus for albuminuria.49 This finding is interesting given the role of cubulin and megalin in the proximal tubule endocytosis processes of numerous proteins. Mechanistically, deficiency in functional cubulin would reduce the reabsorption of albumin from the urine ultrafiltrate through the usual interactions of cubilin with megalin and albumin.50 Supportive animal models have shown a 6-fold increase in albuminuria in cubulin genetic deletion models.51 The missense variant of interest is in a location that is important for the megalin–cubilin interaction.52 The findings in the recent genetics study demonstrating association between the CUBN variant (rs1801239) and urinary albumin excretion were present in both African Americans and patients of European ancestry.53 Each variant copy was associated with a ∼42% increased risk for microalbuminuria at long-term follow-up.53 However, the CUBN variant explains only 0.06–0.15% of the total variance in urinary albumin excretion. A recently published editorial suggests more extensive sequencing of the CUBN gene to evaluate for rarer variants that have larger effects.54 The implications of variants in CUBN on urinary elimination of drugs that are highly bound to albumin are currently unknown.
Nonmuscle Myosin Heavy Chain
The nonmuscle myosin protein is expressed in the podocytes of the glomerulus. Other key proteins including nephrin, CD2AP, podocin, and alpha-actinin 4 are also expressed within the podocytes and their genetic variants are being evaluated in various forms of glomerulonephritis (described later in this chapter). Podocytes are outcroppings from the glomerular epithelial membrane that form the so-called foot processes visualized on microscopy. Podocytes help to form the glomerular slit pores that are important for sieving, for example, large molecules are retained in the blood and small molecules are passed into the glomerular filtrate to the proximal tubule. Additionally, podocytes are involved in regulation of the glomerular filtration rate (GFR) through their ability to contract. Several investigators have found associations between polymorphisms in the MYH9 gene (encoding nonmuscle myosin heavy chain) and kidney disease in African Americans.55–58 More specifically, associations were found with albuminuria in hypertensives56 and idiopathic and HIV-associated focal glomerulosclerosis57 and with end-stage renal disease (ESRD) in hyper-tensives.55,58 The investigators found an odds ratio of 4–5 for focal segmental glomerulosclerosis (FSGS) and HIV nephropathy and 1.5–3.4 for hypertensive ESRD in patients with MYH9 variants.55,57 Variants in MYH9 associated with nephropathy include rs4821480 (T36695247G), rs2032487 (T36695428C), rs4821481 (T36695942C), and rs3752462 (C36710183T). The GCCT haplotype, derived from the SNPs consecutively listed in the previous sentence, is recognized as the “at-risk” haplotype for nephropathy.57 While the association of MYH9 variants with numerous human kidney diseases is promising research, there is still some concern by scientists given that deletion of the MYH9 gene in mice has not been found to elevate the risk of CKDs, with the exception of nephropathy induced by Adriamycin.59
Renin-Angiotensin-Aldosterone System
Medications aimed at the angiotensin pathway, for example, ACEIs and/or ARBs, have been the cornerstone of treatment for the primary and secondary prevention of CKD, especially in diabetic patients. These therapies have shown positive results in preventing and reducing albumin excretion, slowing kidney disease progression, and reducing the risk for doubling of serum creatinine concentrations and/or requiring dialysis or transplantation. Three decades ago, it was reported that the insertion/ deletion (I/D) polymorphism in the angiotensin-converting enzyme (ACE) gene explained ∼50% of the variability in ACE levels.60 This variant is located in intron 16 and is defined by a 250 base pair fragment (rs1799782). Since this initial report, numerous studies have sought to evaluate the influence of the I/D polymorphism on the responses to ACE inhibitors, with findings that have been variable from study to study. A meta-analysis published in 2005 reviewed the response data from 11 studies employing ACEIs.61 While four of the studies evaluated outcomes of one or more kidney-related clinical parameters (proteinuria, serum creatinine, GFR, ESRD), two of them provided the majority of included data.62,63 The study by Perna et al. included 212 nondiabetic, proteinuric, Caucasians who received ramipril versus placebo and were categorized as ACE D/D, D/I, and I/I.63 Reductions in proteinuria, GFR, and albumin excretion rate at 30 months were reported. The outcomes favored the D/D genotype, with D/I genotypes demonstrating a response that was between the D/D and I/I genotypes. The overall effect showed a mean ± SD of 0.33 ± 1.2 g per day reduction in proteinuria and 0.08 ± 0.63 mL/min per month increase in GFR. There was also a 0.65 risk reduction in ESRD that favored the D/D genotype.63 Penno et al. evaluated proteinuria in 530 Caucasian diabetics with normoalbuminuria or microalbuminuria who received lisinopril.62 The study outcome favored the I/I genotype, with a 7.33 μg/min reduction in albumin excretion rate.62 A previous genetic study from 1,365 Caucasian patients from the DCCT and EDIC studies showed that patients with the I/I genotype had a lower risk for persistent microalbuminuria and severe nephropathy.64 More recent studies in Asians reported a benefit of the ACE I/I genotype, lower risk of kidney disease progression from diabetes,65 more decreases in proteinuria and amelioration of kidney function loss with ARB treatment,66 and reduced risk of nephropathy from type II diabetes.67 Two other studies fail to support any association between ACE I/D polymorphism and susceptibility to immunoglobulin A (IgA) nephropathy or autosomal dominant polycystic kidney disease (ADPKD).68,69 The results from the existing studies suggest race-and disease-related differences in kidney outcomes according to ACE I/D genotypes, suggesting the interplay between genetic and nongenetic factors. There is currently no consensus regarding ACEI therapy or dose selection based on the ACE I/D polymorphism in terms of nephropathy reduction.
There have been investigations into the role of polymorphisms in angiotensinogen (AGT M235T, rs699), angiotensin II receptor type I (AGTR1 A1166C, rs5186), angiotensin II receptor type II (AGTR2 A1818T, rs5978731), and aldosterone synthase (CYP11B2 C-344T, rs1799998) and kidney diseases and/or response to treatments employing ARBs. Type I diabetics with the AGTTT/T genotype (at base position 235) had a 4-fold increased risk for microalbuminuria as compared with M/M and M/T genotypes.70 A study of nearly 900 Mexican Americans reported an association between the AGT T/T genotype and reductions in GFR.71 In 239 Asian diabetic patients with proteinuria receiving valsartan, the AGTR1 A1166C homozygous wild-type genotype was associated with a 46% reduction in proteinuria from baseline as opposed to an 11% reduction in patients with the C/C genotype.72 The AGTR2 A1818T genetic variant has been associated with the progression of IgA nephropathy in Koreans.73 A study in Caucasian hypertensive patients reported a possible interaction of the CYP2B11 C/C genotype with the AGT T/T, AGTR1 A/C, and ACE D/D genotypes for the development of kidney insufficiency.74 These data show that polymorphisms in genes within the RAAS pathway other than ACE are being investigated for their potential role in kidney disease, as well as in explaining response to therapies employing ACEIs or ARBs. Evolving data may enable more individualized selection of therapeutic agents in patient with urinary protein excretion.
CYP2C9 Polymorphisms and Losartan Response
Polymorphisms in the cytochrome P450 2C9 gene have been explored for relationships to blood pressure lowering and influence on proteinuria reduction in patients receiving the ARB, losartan. Losartan is a therapeutically active drug that is further metabolized to E3174, an active metabolite. Reductions in metabolic activity of CYP2C9 would be predicted to reduce the overall pharmacodynamic responses secondary to shunting of metabolism to inactive moieties via other metabolism routes. Thus, while all patients would be exposed to the parent drug, exposure to E3174 would be greatest in patients without alterations resulting in the decreased activity of CYP2C9. CYP2C9 *2 (Arg144Cys, rs1799853) and CYP2C9 *3 (Ile359Leu, rs1057910) variants result in a reduced CYP2C9 activity phenotype in patients. However, since the variants are rare in African American patients and more common in Caucasians, their influence would be relevant to blood pressure (systolic and diastolic) and proteinuric responses in the latter group. The influence of CYP2C9 genotype on losartan pharmacodynamic responses was recently evaluated in Caucasian patients with primary and secondary kidney diseases.75 Patients with primary kidney diseases who carried variant alleles had less favorable antiproteinuric responses (31% vs. 125% reductions, respectively). Patients with secondary kidney diseases who carried variant alleles had less favorable reductions in diastolic and systolic blood pressures.75 Lajer et al. evaluated blood pressure responses according to CYP2C9 genotype in Caucasian type I diabetic nephropathy patients receiving losartan.76 Diabetic nephropathy would be considered as an etiology for secondary kidney disease. A significant reduction in systolic blood pressure in the non-*3 carriers versus *3 carriers was noted. No differences in diastolic blood pressure or urinary albumin excretion were noted.76 These two studies suggest reduced pharmacodynamic responses to losartan in Caucasian patients with nephropathy. Since these former studies were of only 4–6 months in duration, the long-term outcomes to losartan therapy based on CYP2C9 genotype are unknown.
GLOMERULONEPHRITIS
Glomerulonephritis is the third leading cause of CKD in the United States and is ranked as the second leading cause in most Asian populations.1
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


