Fig. 11.1
Complexity of ETS gene fusions in prostate cancer. a Multiple 50 partners ( red) and ETS genes ( blue) have been identified. b Two genomic mechanisms of TMPRSS2:ERG gene fusions have been identified. The prostate-specific androgen-induced transmembrane protease serine 2 gene, TMRPSS2, and the v-ets erythroblastosis virus E26 oncogene homolog gene, ERG, are located approximately 3 megabases (Mb) apart on chromosome 21, and fusion can occur either through deletion of the intervening genomic region ( arrows) or insertion of the intervening region to another chromosome. Stylized results obtained by fluorescence in situ hybridization (FISH) using probes located 50 ( green) and 30 ( red) to ERG are shown to the right of the structural diagrams, with colocalization of 50/30 ERG probes indicated in yellow. c Multiple fusion transcript isoforms have been characterized. Stylized structures for TMPRSS2 ( red) and ERG ( blue) are shown. Noncoding and coding exons are shown in small and large boxes, respectively. Transcripts differ in the location of the junction between the 50 partners and the ETS gene, as well as the included exons. d Localized prostate cancer is commonly multifocal, with several distinct appearing foci of cancer. A single prostate can contain foci without ETS gene rearrangements, foci with TMPRSS2:ERG fusion through deletion ( del), or foci with TMRPSS2:ERG fusion through insertion ( ins). (Used with permission from [36])
SPOP Mutations
Mutations in SPOP in prostate cancer have been recently discovered in systematic sequencing studies [2, 7, 19, 39]. These represent the most common point mutations in primary prostate cancer, with recurrent mutations in SPOP in 6–13 % of multiple independent cohorts. The SPOP gene encodes for the substrate-recognition component of a Cullin3-based E3-ubiquitin ligase; missense mutations are found exclusively in the structurally defined substrate-binding cleft of SPOP, indicating that prostate cancer-derived mutations will alter substrate binding [2, 39] (Fig. 11.2a, b).
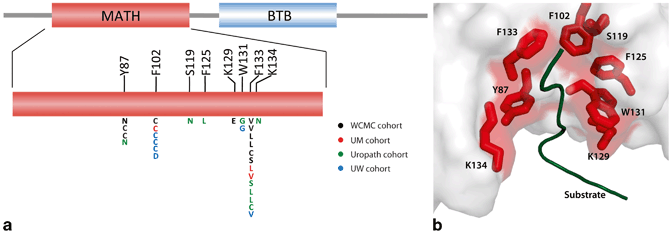
Fig. 11.2
Structural studies of recurrent SPOP alterations in prostate cancer. a Positional distribution of somatic alterations in SPOP across the Weill Cornell Medical College ( WCMC), University of Michigan ( UM), Uropath, and University of Washington ( UW) prostate tumor cohorts. b Mutated residues in the crystal structure of the SPOP MATH domain bound to substrate (PDB 3IVV). MATH meprin and TRAF homology domain, BTB broad complex, tramtrack and bric-a-brac domain. (Used with permission from [2])
Mutations Affecting Gene Expression and Chromatin Regulation
Regulation of chromatin remodeling , the process of modifying DNA architecture through histone modifications and other restructuring processes, has emerged as a major mechanism for alterations across the spectrum of human cancers. Alteration in proteins involved in chromatin regulation can have far reaching cellular effects, affecting genome-wide control of gene expression and playing key roles in DNA repair and genome maintenance. Mutations in a number of genes involved in histone modifications have been identified in prostate cancer. These include KDM6A/UTX, MLL2, and MLL3 [2, 3, 6, 19]. Interestingly, proteins encoded by these genes all act to alter methylation of the histone variant H3, known to be a key component of regulation of chromatin states and involved in transcriptional control.
CHD1 at 5q21 encodes a chromodomain helicase DNA-binding protein that acts to remodel chromatin states (partly by acting as a chaperone of H3.3), and is involved in transcriptional control across the genome. The CHD1 locus is recurrently deleted in prostate cancer, at roughly 10–25 % frequency in both primary and metastatic tumors; rearrangements and point mutations have also been identified [2, 3, 6, 7]. Furthermore, prostate tumors with CHD1 deletion have a significant increase in genomic rearrangements [40]. Future studies will elucidate the role of this putative tumor suppressor in the pathogenesis of prostate cancer.
Enhancer of zeste homolog 2 ( EZH2) acts as a histone methyltransferase (HMT) to silence gene expression and plays a critical role in chromatin regulation. Dysregulation of EZH2 occurs in a variety of human cancers, through mutation, overexpression, and other mechanisms. EZH2 is overexpressed in prostate cancer, and overexpression is associated with aggressive and metastatic disease [41] (Fig. 11.3a, b). Interestingly, recent data show that the role of EZH2 in prostate cancer may be independent of its function in silencing gene expression, but instead it acts as an activator of the AR and other transcription factors [42]. These discoveries raise the possibility that therapeutic targeting of EZH2 activity may be a potential strategy for advanced prostate cancer (see below in epigenetics section).
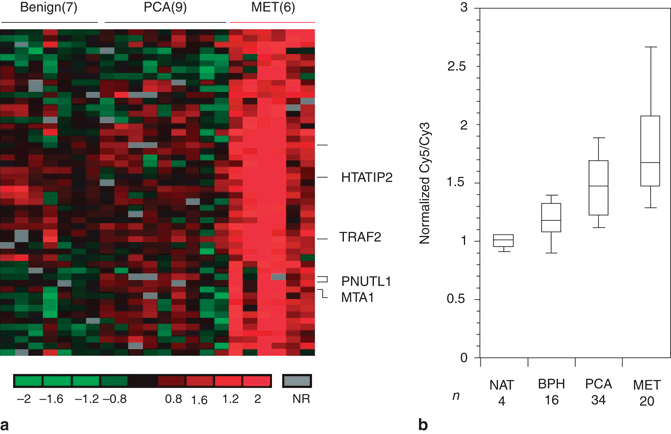
Fig. 11.3
Overexpression of EZH2 in metastatic hormone-refractory prostate cancer ( MET). a Cluster diagram depicting genes that distinguish MET from clinically localized prostate cancer ( PCA). Genes upregulated in METs relative to prostate cancer are shown. Red and green represent upregulation and downregulation, respectively, relative to the median of the reference pool. Grey represents technically inadequate or missing data, and black represents equal expression relative to the reference sample. b DNA microarray analysis of prostate cancer shows upregulation of EZH2 in METs. BPH benign prostatic hyperplasia, NAT normal adjacent prostate tissue. (Used with permission from [41])
Prognostic Significance of Genetic Changes
Although we have begun to catalogue the alterations in prostate cancer, the prognostic significance of the majority of these changes remains unclear. The long natural history of prostate cancer complicates establishing predictive relationships, and raises the possibility that many mutations that drive tumorigenesis in the prostate are not associated with disease progression or mortality. Instead, lesions that initiate cancer may occur decades before the disease becomes clinically relevant, and may have no effect on prognosis. Furthermore, long follow-up on large well-annotated cohorts are necessary to establish effects on prognosis.
PTEN
Dysregulation of PTEN is the lesion most consistently associated with poor prognosis in prostate cancer. A preponderance of evidence shows that deletion of PTEN is associated with advanced localized or metastatic disease, higher Gleason grade , and higher risk of progression, recurrence after therapy, and death from disease [8, 43–47].
TMPRSS2-ERG
As the most common event in prostate cancer, numerous studies have investigated the effect of TMPRSS2-ERG fusion on prognosis. Data are conflicting; ETS fusions have been reported as associated with both more aggressive and more indolent disease, likely representing heterogeneity of study cohorts and management, the impact of sampling, multifocality and intraprostate molecular heterogeneity, and the variability of measured outcomes. Here we will discuss briefly what Tomlins et al. have reviewed in details [36]. Population-based studies focused on non-PSA screened populations with prostate cancer diagnosed by transurethral resection of the prostate (TURP) and conservatively managed (watchful waiting) have shown a significant association between ERG rearrangement and adverse clinicopathologic predictors, metastases, or disease-specific death [48, 49]. Studies investigating the impact of ETS fusions on aggressive features or outcome following radical prostatectomy have produced conflicting results, with several showing association between ETS fusion status and features of aggressive prostate cancer (including increased Gleason grade, stage, or biochemical recurrence [BCR]), while others have found no such associations, or even the opposite (association with lower Gleason grade or increased recurrence-free survival). In summary, population-based studies of watchful waiting cohorts have shown ETS fusions associated with poor prognosis, while retrospective radical prostatectomy series have conflicting results regarding aggressiveness and prognosis of ETS fusion-positive cancers; variation in techniques to detect ERG rearrangement also confounds interpretation across studies.
Somatic Copy Number Alterations (SCNAs) and Gene Expression
In addition to the effect of specific genomic events on the prognosis of prostate cancer, the implications of genome-wide or transcriptome-wide changes have also been investigated. Multiple authors have shown that the overall number of SCNAs correlates with Gleason grade, tumor stage , and other poor prognostic features [6, 15, 50]. This may reflect the impact of the overall degree of genomic instability in these tumors, or may represent the accumulation of driving events, with prognosis worsening as the tumor accumulates additional “hits.” Studies investigating gene expression have been also attempted to define patterns associated with aggressive disease; many studies have reported gene expression signatures predictive of disease progression or aggressiveness, but limited value has been demonstrated across cohorts and transition to the clinical setting remains elusive.
Tumor Heterogeneity and Potential Targets
The heterogeneity of prostate cancer complicates risk stratification and selection of management strategies. However, molecular classification holds the promise of identifying specific subclasses of prostate cancer associated with distinct patterns of genomic abnormalities. Genomic and transcriptomic analyses reveal that prostate tumors can be subclassified based on gene expression and SCNA signatures, with some success in predicting aggressive features of disease or impact on prognosis [6, 15, 50, 51]. Systematic sequencing studies continue to add data allowing the definition of molecular subclasses based on mutations and copy number aberrations. These discoveries raise the possibility that prostate cancer might soon transition from a poorly understood, clinically heterogeneous disease to a collection of homogenous subtypes identifiable by molecular criteria , associated with specific genetic abnormalities, with distinct effects on patient prognosis, amenable to specific management strategies, and perhaps vulnerable to specific targeted therapies. As these subclasses emerge, selection of model systems based on genetic context becomes critical; for instance, studying SPOP mutations in a cell line that has TMPRSS2-ERG fusion or TP53 mutations may be futile, since these events are mutually exclusive in tumors.
ETS Fusion-Positive Tumors
Due to the approximately 50 % prevalence of ETS fusions, attempts to molecularly characterize prostate often begin with division into ETS-positive and ETS-negative subclasses. It is likely that the different ETS fusions genes have similar functional consequences to the cancer cell. Although prostate tumors have been reported with more than one type of ETS fusion, in general only a single ETS fusion is present in a given tumor, consistent with functional redundancy [52]. Multiple studies have defined distinct gene expression profiles in ETS fusion-positive and ETS fusion-negative prostate cancers [36, 51, 53]. In addition, tumors with ERG rearrangement have distinct SCNA profiles and increased lesions in TP53 and PTEN, suggesting that they represent a biologically distinct entity [2, 6, 54].
The high prevalence and simple identification of prostate cancers with ETS rearrangement led to interest in potential therapeutic targeting. Although successful targeted therapy against oncogenic transcription factors has proven notoriously difficult, Brenner et al. identified the enzyme poly (ADP-ribose) polymerase 1 (PARP1) as an ERG-interacting protein critical for the oncogenic action of ETS proteins in prostate cancer cells, and demonstrated that inhibition of PARP resulted in decreased growth of ETS fusion-positive, but not ETS-negative prostate cancer xenografts [55]. These findings suggest that PARP inhibitors, currently under clinical investigation in a number of cancers, including breast and ovarian, represent a potential therapeutic avenue specifically for ETS-positive prostate cancers.
SPOP Mutations Define a Distinct Molecular Class of Prostate Cancer
Mutations in SPOP occur in up to 15 % of prostate cancers; importantly, SPOP mutations are mutually exclusive with TMPRSS-ERG fusion and other ETS rearrangements, and SPOP-mutant tumors generally lack lesions in the PI3K pathway [2, 3, 19] (Fig. 11.4). Moreover, SPOP mutations are also mutually exclusive with deletions and mutations in the TP53 tumor suppressor [2, 19]. Finally, SPOP-mutant tumors show a distinct pattern of genomic aberrations; specifically, deletions of CHD1 at 5q21.1 and deletion in the 6q21 region are significantly associated with SPOP mutations [2]. Taken together, these findings support SPOP mutations as a driver lesion that underlies a distinct molecular subclass of prostate cancer.
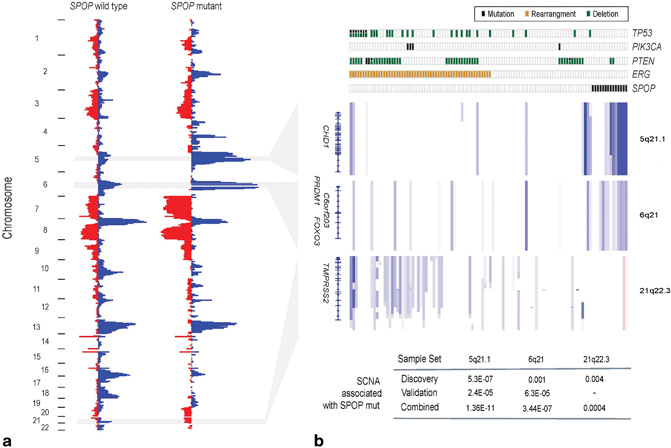
Fig. 11.4
SPOP mutation defines a distinct genetic subclass of prostate cancer. a Heat map showing selected recurrent somatic copy-number aberrations (SCNAs). b Each column represents a single prostate cancer sample. Samples are annotated for mutations in SPOP, PTEN, PIK3CA and TP53, deletions of PTEN, and ERG rearrangements. Deletions positively correlated (5q21.1, 6q21) or inversely correlated (21q22.3) with SPOP mutation are shown. P values of peak association with SPOP mutation in both discovery and validation cohorts are given below (Fisher’s exact test). (Used with permission from [2])
SPINK1
As studies have characterized the molecular nature of prostate cancer, additional potential subtypes have emerged. The serine peptidase inhibitor, Kazal type 1 ( SPINK1) is a secreted protein overexpressed specifically in a subset of ETS-negative cancers [56–58]. SPINK1 overexpression is associated with decreased BCR-free survival, and monoclonal antibodies to SPINK1 attenuate the growth and invasion of SPINK1-positive cells in prostate cancer models. Furthermore, EGFR, through interaction with SPINK1, may in part mediate the oncogenic effects of SPINK1, and inhibition of EGFR signaling with already clinically established agents may be another route of targeted therapy for this specific subclass of prostate cancer [59]. These studies on ETS fusion and SPINK1-positive prostate cancer subclasses can serve as a model for how further classification efforts can benefit patients; identifying molecular subclasses with specific underlying genetic abnormalities, finding effects on patient prognosis, defining the signaling pathways associated with these lesions that may drive prostate tumorigenesis, and identifying potential targets for therapy.
IL-6 and Cytokine Signaling
Accumulating evidence also implicates cytokine signaling as a targetable axis in prostate cancer. Interleukin-6 (IL-6) is an inflammatory cytokine that is overexpressed in prostate cancer; it regulates proliferation, apoptosis, and angiogenesis through activation of multiple downstream pathways, including MAPKs and Akt. While no specific mutations in elements of IL-6 signaling have been reported, preclinical studies in multiple prostate tumor models reveal the potential of the anti-IL-6 antibody siltuximab, and clinical trials have been initiated [60]. Endogenous inhibitors of cytokine signaling are also relevant in prostate cancer; Suppressor of Cytokine Signaling 3 ( SOCS3) inhibits apoptosis in AR-negative models [61]. IL-6 has pleomorphic effects that are cell-context dependent, complicating the search for biomarkers and design of trials [62].
Rare Lesions and Opportunities for Precision Medicine
The high incidence of prostate cancer and the diverse and heterogeneous pattern of alterations in the disease imply that even alterations only impacting a few percent of patients may have clinical utility; this is the paradigm of personalized medicine. Highlighting this are recent studies identifying rare fusion genes involving the Ras/Raf kinase pathways. Rearrangements of the BRAF or RAF1 genes have been reported in 1–2 % of prostate cancers [9, 17] (Fig. 11.5a–d), while KRAS rearrangement has also been discovered in advanced prostate cancer [18]. Importantly, activating events in these pathways are considered targetable by existing Raf kinase inhibitors. These studies suggest that while uncommon, such events may define a model where evaluation of the molecular profile of an individual’s prostate cancer could reveal rare but actionable alterations that can be treated with existing pharmacologic agents.
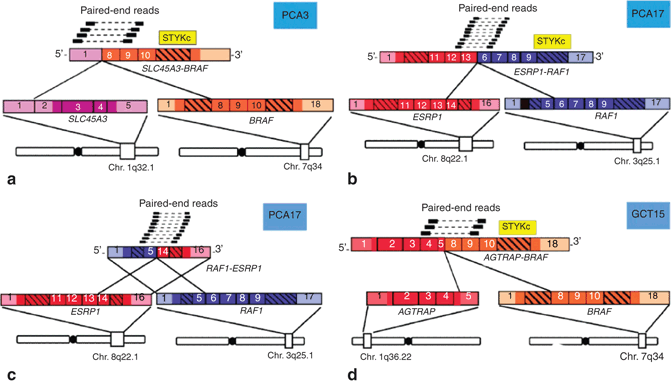
Fig. 11.5
Discovery of the SLC45A3-BRAF and ESRP1-RAF1 gene fusions in prostate cancer by paired-end transcriptome sequencing. a Schematic representation of reliable paired-end reads supporting the interchromosomal gene fusion between SLC45A3 ( purple) and BRAF ( orange). The protein kinase domain in the BRAF gene ( yellow) remains intact following the fusion event. Respective exons are numbered. b, c As in a, except showing the fusions between ESRP1 ( red) and RAF1 ( blue), resulting in reciprocal fusion genes ESRP1-RAF1 and RAF1-ESRP1. d As in a, except showing the fusion between AGTRAP ( red) and BRAF ( orange). (Used with permission from [17])
A subtype of prostate cancer that can rarely arise de novo, neuroendocrine prostate cancer (NEPC), is a lethal subtype that most commonly develops after hormonal therapy for the disease [63]. It is estimated that up to 30 % of late stage prostate cancers harbor a predominance of neuroendocrine differentiation. NEPC does not secrete PSA or express AR, and should be suspected in patients with rapid disease progression especially metastases to visceral organs, low or modestly elevated serum PSA level and elevated serum markers of neuroendocrine differentiation (i.e., chromogranin A or neuronspecific enolase). Recent transcriptome sequencing and assessment of DNA copy number changes of both prostate cancer and NEPC has brought new insight into NEPC pathogenesis [9]. Despite clonal origin of NEPC from adenocarcinoma cells, there exist dramatic gene expression differences with nearly 1000 genes showing differential expression. In addition, the genome of NEPC is widely aberrant with frequent amplifications and deletions. There are subpopulations of prostate cancer patients that demonstrate mixed molecular features and may be at high risk for progression to NEPC. For instance, co-amplification of the genes encoding the oncogenes Aurora kinase A (AURKA) and N-myc ( MYCN) are frequently found in primary tumors of patients that later develop treatment-related NEPC, and are infrequent in other primary prostate adenocarcinomas [64]. Therefore, AURKA and MYCN amplifications may predict patients at high risk for the development of treatment-related NEPC.
Temporal Relationships Among Genomic Events
Establishing the temporal sequence of genomic events in prostate cancer—which lesions occur early and likely initiate cancer, versus those that come later and are associated with disease progression—is critical for defining prostate cancer progression and aggressiveness at the molecular level. A molecular definition of progression may be invaluable for patients on active surveillance or for risk-stratification of intermediate-risk patients. ERG rearrangement has been shown in both isolated high-grade prostatic intraepithelial neoplasia (HGPIN) and in HGPIN adjacent to invasive prostate cancer [36, 65]. SPOP mutations have also been identified in HGPIN, and are only observed in ETS-negative tumors, suggesting that SPOP mutation and ETS rearrangements are mutually exclusive early events in the natural history of prostate cancer [2]. In contrast, lesions in PTEN, RB1, TP53, and AR are more commonly reported in advanced tumors. Whole genome sequencing has provided additional insight in this endeavor. Analysis of the clonality of genomic events (in essence, the percentage of cells in a tumor with a specific lesion) allows investigators to extrapolate the hierarchy of these events in a tumor’s natural history. Using this approach, Baca et al. have reported ERG rearrangement, NKX3-1 deletion, and mutations in SPOP and FOXA1 as clonal, early events in the history of prostate cancer. These are followed by lesions in CDKN1B and TP53, and finally by inactivation of PTEN [40] (Fig. 11.6). Findings such as these establish a framework for defining the sequence of molecular events in the natural history of prostate cancer, from disease initiation to progression, metastases, emergence of treatment resistance, and death.
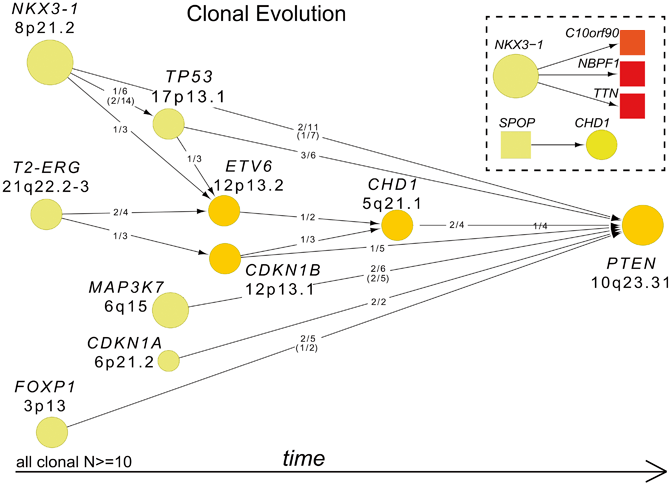
Fig. 11.6
Clonality and evolution of prostate cancer. Patterns of tumor evolution were inferred on the basis of clonality estimates. Arrows indicate the direction of clonal–subclonal hierarchy between genes that are deleted in the same sample in multiple cases. Deleted genes are represented by circles with size and color intensity reflecting the frequency of overall deletions and subclonal deletions, respectively. Ratios along the arrows indicate the number of samples demonstrating directionality of the hierarchy out of samples with deletion of both genes. (Used with permission from [40])
Section Summary
Major advances have been made in cataloguing the genomic alterations in prostate cancer, understanding the molecular mechanisms underlying the disease, and using this information to subclassify tumors. These findings raise the possibility that prostate cancer could soon transition from a poorly understood, heterogeneous disease with a variable clinical course to a collection of homogenous subtypes, identifiable by molecular criteria, associated with distinct risk profiles, and perhaps amenable to specific management strategies or targeted therapies [66] (Fig. 11.7).
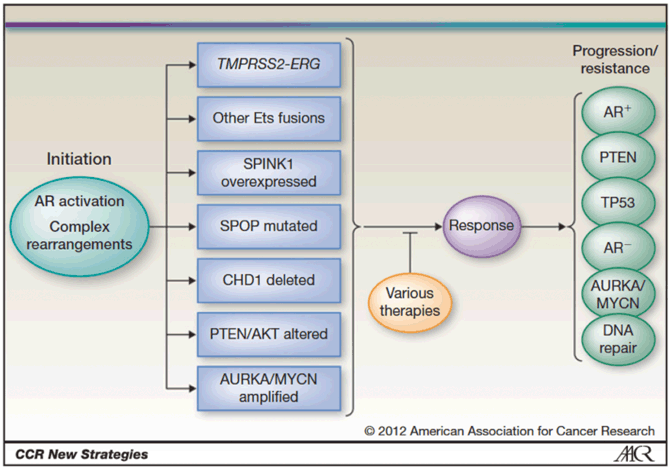
Fig. 11.7
Molecular subclassification of prostate cancer. Disease initiation occurs through activation of androgen signaling, complex rearrangements, or other proposed mechanisms, leading to prostate tumor—wide clinical heterogeneity and distinct molecular subtypes. Disease progression and resistance to therapy lead to acquisition of new and potentially overlapping molecular alterations. (Used with permission from [66] Copyright © American Association for Cancer Research.)
Epigenetic Alterations in Prostate Cancer
Epigenetic alterations occur more frequently than somatic mutations . They arise early and associate with prostate cancer and disease progression, including DNA methylation , histone modifications , and microRNA (miRNA) regulation . These heritable alterations affect gene expression without changing the DNA sequences during the development of prostate cancer. As epigenetic alterations can be reversed, inhibitors blocking the processes are being tested for their effectiveness in treating prostate cancer. In addition, there appear to be many opportunities for biomarker development based on epigenetic alterations as these changes can be robustly measured.
DNA Methylation
GSTP1
The best-known gene-specific hypermethylation in prostate cancer is Glutathione-S-transferase P1 ( GSTP1) [67]. Loss of expression of GSTP1 is frequently found in prostate cancer, as well as in HGPIN, and its silencing is a result of promoter hypermethylation. These observations support that promoter hypermethylation is an early event in tumorigenesis . GSTP1 is involved in oxidative damage response and can protect cells from DNA damage and cancer initiation. Lack of GSPT1 expression might enable cells to tolerate DNA damage and mutations. Hypermethylation at the GSTP1 promoter in prostate cancer is well characterized and documented. Since it can be detected in tumor tissues and body fluids (urine and blood), extensive work has been done in an attempt to make GSTP1 a useful biomarker. When screening for GSTP1 promoter hypermethylation, it shows high specificity, but various sensitivities in urine and serum/plasma. The discrepancies among different studies reflect the differences in methodologies and patient cohorts. Detecting GSTP1-promoter hypermethylation has been suggested as biomarker for diagnosis, prognosis, and treatment response . For example, serum GSTP1 promoter hypermethylation has been suggested as a predictor of BCR in patients with localized disease at radical prostatectomy . Still, some prostate cancer cases do not have GSTP1 promoter hypermethylation, and examination of panels of multiple genes has been proposed to balance this scenario. Combination of multiple genes has greatly enhanced the detection while maintaining high specificity; gene selection is critical and should be carefully evaluated. In a recent large-scale study, the panel of GSTP1, HIF3A, HAAO, and RARβ provided high sensitivity and high specificity to distinguish prostate cancer from benign prostate tissues [68] .
Genome-Wide DNA Methylation Profiling
In the last decade, our understanding of DNA methylation changes in prostate cancer have been greatly improved, thanks in part to new technologies (e.g., microarray and Next Generation Sequencing). The DNA methylation patterns in prostate cancer show global losses with a particular enrichment at CpG island promoter regions. Regions with medium to low CpG density tend to develop DNA hypomethylation during prostate cancer initiation. While next generation sequencing allows for deeper coverage, new understanding of differentially methylated regions and their associated genes are being identified. Analyses integrating mutations, copy number variation, and gene expression data can further provide relevant information to the cancer genome. Different from candidate gene-based approaches, genome-wide site-specific DNA methylation events can now be studied and previously unknown differential methylation regions can be identified to provide a more complete map of DNA methylation changes.
Methylation profiling with microarray technology is limited by defined probes; however, several studies have uncovered biomarkers for predicting BCR [69], associated with prostate cancer progression [70, 71] (Fig. 11.8a–c), and the combination of methylation and copy number changes has shown that in CRPC copy number loss and promoter hypermethylation occur together for tumor suppressor genes (e.g., RB) in the same tumor [72]. These results suggest that multiple mechanisms are utilized simultaneously by cancer cells to gain growth advantages.
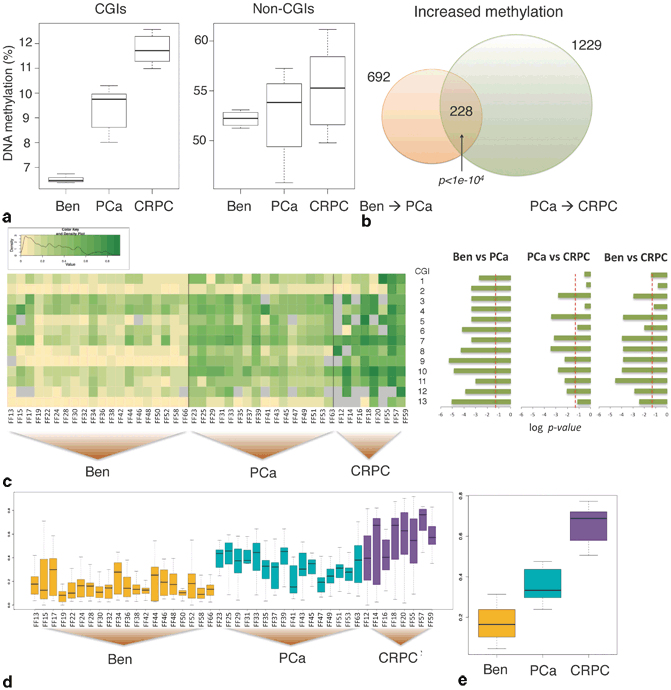
Fig. 11.8




DNA methylation increases with and may predict disease severity. a Left: Heat map of DNA methylation levels of the panel of 13 CpG islands ( CGIs) in the three groups, 20 benign prostate tissues and 16 PCa and 8 CRPC samples (value range, 0–1). Right: Comparison between the three groups with adjusted log P values (Benjamini-Hochberg correction false discovery rate (FDR) controlled at 0.05). b Boxplots of the average DNA methylation levels of the panel for individual samples. c Boxplots of the average DNA methylation levels of the panel for the three groups. The AUC, sensitivity, and specificity for the comparison of benign and PCa are 0.9375, 100, and 75 %, and those for the comparison of PCa and CRPC are 0.975, 95, and 95 %. The boxplots at the bottom show average DNA methylation levels in individual samples (d) and in the three groups (e). (Used with permission of [71])
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
