General Principles of Antimicrobial Therapy
ANTIMICROBIAL CHEMOTHERAPY
Microorganisms of medical importance fall into 4 categories: bacteria, viruses, fungi, and parasites. Likewise, antibiotics are broadly classified as (1) antibacterial, (2) antiviral, (3) antifungal, and (4) antiparasitic agents. Antimicrobial molecules should be viewed as ligands whose receptors are microbial proteins. The microbial proteins targeted by the antibiotic are essential components of biochemical reactions in the microbes, and interference with these physiological pathways kills the microorganisms. The biochemical processes commonly inhibited include cell wall synthesis in bacteria and fungi, cell membrane synthesis, synthesis of 30s and 50s ribosomal subunits, nucleic acid metabolism, function of topoisomerases, viral proteases, viral integrases, viral envelope fusion proteins, folate synthesis in parasites, and parasitic chemical detoxification processes.
Classification of an antibiotic is based on:
• The class and spectrum of microorganisms it kills
• The biochemical pathway it interferes with
• The chemical structure of its pharmacophore
The relationship between antimicrobial concentration and effect on a population of organisms is modeled using the standard Hill-type curve for receptor and agonist (see Chapters 2 and 3), characterized by 3 parameters: the inhibitory concentration 50, or IC50 (also termed EC50), a measure of the antimicrobial agent’s potency; the maximal effect, Emax; and H, the slope of the curve, or Hill factor. In antimicrobial therapy, the relationship is often expressed as an inhibitory sigmoid Emax model, to take into account the control bacterial population without treatment (Econ) as a fourth parameter (Equation 48–1 and Figure 48–1), where E is the effect as measured by microbial burden.
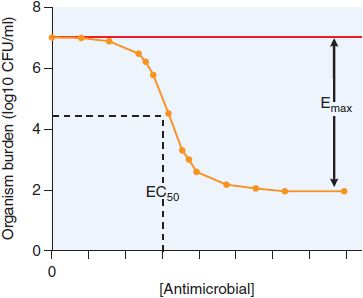
Figure 48–1 Inhibitory sigmoid Emax curve. CFU, colony-forming unit.
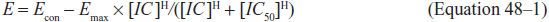
THE PHARMACOKINETIC BASIS OF ANTIMICROBIAL THERAPY
PENETRATION OF ANTIMICROBIAL AGENTS INTO ANATOMIC COMPARTMENTS. In many infections, the pathogen causes disease not in the whole body, but in specific organs. Antibiotics are often administered far away from these sites of infection. Therefore, in choosing an antimicrobial agent for therapy, a crucial consideration is whether the drug can penetrate to the site of infection.
For example, the antibiotic levofloxacin achieves skin tissue/plasma peak concentration ratio of 1.4, epithelial lining fluid to plasma ratio of 2.8, and urine-to-plasma ratios of 67. In clinical trials with levofloxacin, the failure rate of therapy was 0% in patients with urinary tract infections, 3% in patients with pulmonary infections, and 16% in patients with skin and soft tissue infections. Clearly, the poorer the penetration into the anatomical compartment, the higher the likelihood of failure. Penetration of a drug into an anatomical compartment depends on the physical barriers that the molecule must traverse, the chemical properties of the drug, and the presence of multidrug transporters. The physical barriers are usually due to layers of epithelial and endothelial cells, and the type of junctions formed between these cells. Penetration across this physical barrier generally correlates with the octanol-water partition coefficient of the antimicrobial agent, a measure of its hydrophobicity. Hydrophobic molecules get concentrated in the cell membrane bilayer, whereas hydrophilic molecules tend to concentrate in the blood, the cytosol, and other aqueous compartments (see Figure 2–3).
Figure 48–3 Changes in sigmoid Emax model with increases in drug resistance. Increase in resistance may show changes in IC50 (panel A: the IC50 increases from 70 [orange line] to 100 [green line], to 140 [blue line]) or decrease in Emax (panel B: efficacy decreases from full response [orange line] to 70% [green line]).
Membrane transporters comprise another barrier; they actively export drugs from the cellular or tissue compartment back into the blood (see Chapter 5). A well-known example is the P-glycoprotein. Although the octanol-water partition coefficient would favor lipophilic molecules to transverse across cell barriers, P-glycoprotein exports structurally unrelated amphiphilic and lipophilic molecules of 3-4 kDa, reducing their effective penetration. Antimicrobial agents that are P-glycoprotein substrates include HIV protease inhibitors, the antiparasitic agent ivermectin, the antibacterial agent telithromycin, and the antifungal agent itraconazole.
The CNS is guarded by the blood-brain barrier. The movement of antibiotics across the blood-brain barrier is restricted by tight junctions that connect endothelial cells of cerebral micro-vessels to one another in the brain parenchyma, as well as by protein transporters. Antimicrobial agents that are polar at physiological pH generally penetrate poorly; some, such as penicillin G, are actively transported out of the cerebrospinal fluid (CSF) and achieve CSF concentrations of only 0.5-5% of that achieved in plasma. However, the integrity of the blood-brain barrier is diminished during active bacterial infection; tight junctions in cerebral capillaries open, leading to a marked increase in the penetration of even polar drugs.
The eye, the epithelial lining fluid of the lung, and biofilms and vegetations on artificial heart valves and indwelling catheters pose special problems for drug penetration and effective therapy.
PHARMACOKINETIC COMPARTMENTS. Once an antibiotic has penetrated to the site of infection, it may be subjected to processes of elimination and distribution that differ from those in the blood. These sites where the concentration-time profiles differ from each other are considered separate pharmacokinetic compartments, thus, the human body is viewed as multicompartmental. The concentration of antibiotic within each compartment is assumed to be homogeneous. The model is also defined as open or not open; an open model is one in which the drug is eliminated out of the body from the compartment (e.g., kidneys). The order of the process must also be specified (see Chapter 2): a first-order process is directly correlated to concentration of drug D, or [D]1, as opposed to zero order, which is independent of [D] and reflects a process that is saturated at ambient levels of D.
Suppose a patient has pneumonia with the pathogen in the lung epithelial lining fluid (ELF). The patient ingests an antibiotic that is absorbed via the GI tract (g) into blood or central compartment (compartment 1), as a first-order input. In this process, the transfer constant from the GI tract to central compartment is termed the absorption constant and is designated ka. The antibiotic in the central compartment is then delivered to the lungs where it penetrates into the ELF (compartment 2). However, it also penetrates into other tissues of the body peripheral to the site of infection, termed the peripheral compartment (compartment 3). Thus, we have 4 compartments (including g, a specific compartment, the GI tract, from the set of initial absorption compartments labeled “p” in Figure 48–2), each with its own concentration-time profile, as shown in Figure 48–2. The penetration of drug from compartment 1 to 2 is based on the penetration factors discussed earlier and is defined by the transfer constant k12. However, the drug also redistributes from compartment 2 back to 1, defined by transfer constant k21. A similar process between the blood and peripheral tissues leads to transfer constants k13 and k31. The drug may also be lost from the body (i.e., open system) via the lungs and other peripheral tissues (e.g., kidneys or liver) at a rate proportional to the concentration.
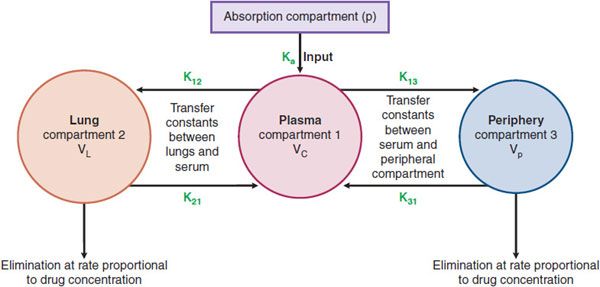
Figure 48–2 Diagrammatic depiction of a multicompartment model.
Antibiotic concentrations within each compartment change with time (the changes are described using standard differential equations). If X is the amount of antibiotic in a compartment, SCL the drug clearance, and Vc the volume of central compartment, then equations for absorption compartment (Equation 48–2), central compartment (Equation 48–3), site of infection or compartment 2 (Equation 48–4), and peripheral compartment (Equation 48–5) are as follows:

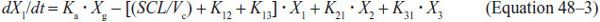


Such models have been used in conjunction with population pharmacokinetics to describe and model a plethora of antimicrobials used to treat bacteria, fungi, viruses, and parasites. Recently, the models have been refined to include sub-populations of the pathogen (killed, inhibited, or resistant to the antibiotic) and other refinements described in Chapter 48 of the 12th edition of the parent text.
POPULATION PHARMACOKINETICS AND VARIABILITY IN DRUG RESPONSE. When multiple patients are treated with the same dose of a drug, each patient will achieve pharmacokinetic parameters that differ from others. This is termed between-patient variability. Even when the same dose is administered to the same patient on 2 separate occasions, the patient may achieve a different concentration-time profile of the drug between the 2 occasions. This is termed inter-occasion or within-patient variability. The variability is reflected at the level of the compartmental pharmacokinetic parameters such as ka, k12, k21, SCL, Vc, and so on. Even when a recommended dose is administered, the drug may fail to reach a therapeutic concentration in some patients. In other patients, the drug may reach high and toxic concentrations. Such variability could be due to genetic variability. In addition, weight, height, and age also lead to variability. Furthermore, patients may have comorbid conditions such as renal and liver dysfunction, which may lead to variability. Drug interactions are an important source of variability with potentially dangerous consequences (see Chapters 5 and 6). Even when such factors are accounted for, there remains residual variability due to computational noise, assay variability, and unexplainable factors. The common practice of using an “average” value of data or “naive pooling” may prevent recognition of subgroups of patients at risk for therapeutic failure or increased toxicity of antibiotic. Knowledge of covariates associated with pharmacokinetic variability leads to better dose adjustments, or switching therapy from one antibiotic to another, or changing concomitant medications.
IMPACT OF SUSCEPTIBILITY TESTING ON SUCCESS OF ANTIMICROBIAL AGENTS
Once the microbial species causing the disease has been identified, a rational choice of the class of antibiotics likely to work in the patient can be made. The microbiology laboratory then plays a second role, which is to perform susceptibility testing to narrow down the list of possible antimicrobials that could be used.
Millions of individuals across the globe get infected by many different isolates of the same species of pathogen. Evolutionary processes cause each isolate to be slightly different from the next, so that each will have a unique susceptibility to antimicrobial agents. As the microorganisms divide within the patient, they may undergo further evolution. Therefore, we expect that there will be a wide distribution of concentrations of antimicrobial agents that can kill pathogens. Often, this distribution is Gaussian, with a skew that depends on where the patient lives. These factors will affect the shape of the inhibitory sigmoid Emax model curve described by Equation 48–1.
With changes in susceptibility, the sigmoid Emax curve shifts in 1 of 2 basic ways. The first is a shift to the right, an increase in IC50, as shown in Figure 48–3A. This means that much higher concentrations than before are now needed to show specific effect. Susceptibility tests for bacteria, fungi, parasites, and viruses have been developed to determine whether these shifts have occurred at a sufficient magnitude to warrant higher doses of drug to achieve particular effect. The change in IC50 may become so large that it is not possible to overcome the concentration deficit by increasing the antimicrobial dose without causing toxicity to the patient. At that stage, the organism is now “resistant” to the particular antibiotic. A second possible change in the curve is a decrease in Emax (Figure 48–3B), such that increasing the dose of the antimicrobial agent beyond a certain point will achieve no further effect; i.e., changes in the microbe are such that eradication of the microbe by the particular drug can never be achieved. This occurs because the available target proteins have been reduced or the microbe has developed an alternative pathway to overcome the biochemical inhibition.
Bacteria. Dilution tests for bacterial drug susceptibility employ antibiotics in serially diluted drug concentrations on solid agar or in broth medium that contains a culture of the test microorganism. The lowest concentration of the agent that prevents visible growth after 18-24 h of incubation is known as the minimum inhibitory concentration (MIC).
Fungi. For fungi that are yeasts (i.e., Candida
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


