General Anesthetics and Therapeutic Gases
General Anesthetics
General anesthetics depress the central nervous system to a sufficient degree to permit the performance of surgery and unpleasant procedures. General anesthetics have low therapeutic indices and thus require great care in administration. The selection of specific drugs and routes of administration to produce general anesthesia is based on their pharmacokinetic properties and on the secondary effects of the various drugs, in the context of the proposed diagnostic or surgical procedure and with the consideration of the individual patient’s age, and associated medical condition.
GENERAL PRINCIPLES OF SURGICAL ANESTHESIA
The administration of general anesthesia is driven by 3 general objectives:
1. Minimizing the potentially deleterious direct and indirect effects of anesthetic agents and techniques
2. Sustaining physiologic homeostasis during surgical procedures that may involve major blood loss, tissue ischemia, reperfusion of ischemic tissue, fluid shifts, exposure to a cold environment, and impaired coagulation
3. Improving postoperative outcomes by choosing techniques that block or treat components of the surgical stress response, which may lead to short- or long-term sequelae
HEMODYNAMIC EFFECTS OF GENERAL ANESTHESIA. The most prominent physiological effect of anesthesia induction is a decrease in systemic arterial blood pressure. The causes include direct vasodilation, myocardial depression, or both; a blunting of baroreceptor control; and a generalized decrease in central sympathetic tone. Agents vary in the magnitude of their specific effects, but in all cases the hypotensive response is enhanced by underlying volume depletion or preexisting myocardial dysfunction.
RESPIRATORY EFFECTS OF GENERAL ANESTHESIA. Nearly all general anesthetics reduce or eliminate both ventilatory drive and the reflexes that maintain airway patency. Therefore, ventilation generally must be assisted or controlled for at least some period during surgery. The gag reflex is lost, and the stimulus to cough is blunted. Lower esophageal sphincter tone also is reduced, so both passive and active regurgitation may occur. Endotracheal intubation has been a major reason for a decline in the number of aspiration deaths during general anesthesia. Muscle relaxation is valuable during the induction of general anesthesia where it facilitates management of the airway, including endotracheal intubation. Neuromuscular blocking agents commonly are used to effect such relaxation (see Chapter 11). Alternatives to an endotracheal tube include a face mask and a laryngeal mask, an inflatable mask placed in the oropharynx forming a seal around the glottis.
HYPOTHERMIA. Patients commonly develop hypothermia (body temperature <36°C) during surgery. The reasons include low ambient temperature, exposed body cavities, cold intravenous (IV) fluids, altered thermoregulatory control, and reduced metabolic rate. Metabolic rate and total body oxygen consumption decrease with general anesthesia by ~30%, reducing heat generation. Hypothermia may lead to an increase in perioperative morbidity. Prevention of hypothermia is a major goal of anesthetic care.
NAUSEA AND VOMITING. Nausea and vomiting continue to be significant problems following general anesthesia and are caused by an action of anesthetics on the chemoreceptor trigger zone and the brainstem vomiting center, which are modulated by serotonin (5HT), histamine, ACh, and dopamine (DA). The 5HT3 receptor antagonists, ondansetron and dolasetron (see Chapters 13 and 46), are very effective in suppressing nausea and vomiting. Common treatments also include droperidol, metoclopramide, dexamethasone, and avoidance of N2O. The use of propofol as an induction agent and the nonsteroidal anti-inflammatory drug ketorolac as a substitute for opioids may decrease the incidence and severity of postoperative nausea and vomiting.
OTHER EMERGENCE AND POSTOPERATIVE PHENOMENA. Hypertension and tachycardia are common as the sympathetic nervous system regains its tone and is enhanced by pain. Myocardial ischemia can appear or worsen during emergence in patients with coronary artery disease. Emergence excitement occurs in 5-30% of patients and is characterized by tachycardia, restlessness, crying, moaning, and thrashing. Neurologic signs, including delirium, spasticity, hyperreflexia, and Babinski sign, are often manifest in the patient emerging from anesthesia. Postanesthesia shivering occurs frequently because of core hypothermia. A small dose of meperidine (12.5 mg) lowers the shivering trigger temperature and effectively stops the activity. The incidence of all of these emergence phenomena is greatly reduced with opioids and α2 agonists (dexmedetomidine).
Airway obstruction may occur during the postoperative period because residual anesthetic effects. Pulmonary function is reduced following all types of anesthesia and surgery, and hypoxemia may occur. Pain control can be complicated in the immediate postoperative period and respiratory suppression associated with opioids can be problematic. Regional anesthetic techniques are an important part of a perioperative approach that employs local anesthetic wound infiltration; epidural, spinal, and plexus blocks; and nonsteroidal anti-inflammatory drugs, opioids, α2 adrenergic receptor agonists, and NMDA receptor antagonists.
ACTIONS AND MECHANISMS OF GENERAL ANESTHETICS
THE ANESTHETIC STATE
The components of the anesthetic state include:
• Amnesia
• Immobility in response to noxious stimulation
• Attenuation of autonomic responses to noxious stimulation
• Analgesia
• Unconsciousness
The potency of general anesthetic agents is measured by determining the concentration of general anesthetic that prevents movement in response to surgical stimulation. For inhalational anesthetics, anesthetic potency is measured in MAC units, with 1 MAC defined as the minimum alveolar concentration that prevents movement in response to surgical stimulation in 50% of subjects. The strengths of MAC as a measurement are that:
• Alveolar concentrations can be monitored continuously by measuring end-tidal anesthetic concentration using infrared spectroscopy or mass spectrometry.
• It provides a direct correlate of the free concentration of the anesthetic at its site(s) of action in the CNS.
• It is a simple-to-measure end point that reflects an important clinical goal.
End points other than immobilization also can be used to measure anesthetic potency. For example, the ability to respond to verbal commands (MACawake) and the ability to form memories also have been correlated with alveolar anesthetic concentration. Verbal response and memory formation are suppressed at a fraction of MAC. The ratio of the anesthetic concentrations required to produce amnesia and immobility vary significantly among different inhalational anesthetic agents (nitrous oxide versus isoflurane).
Generally, the potency of IV agents is defined as the free plasma concentration (at equilibrium) that produces loss of response to surgical incision (or other end points) in 50% of subjects.
MECHANISMS OF ANESTHESIA
CELLULAR MECHANISMS OF ANESTHESIA. General anesthetics produce 2 important physiologic effects at the cellular level:
• The inhalational anesthetics can hyperpolarize neurons. This may be an important effect on neurons serving a pacemaker role and on pattern-generating circuits.
• Both inhalational and IV anesthetics have substantial effects on synaptic transmission and much smaller effects on action-potential generation or propagation.
Inhalational anesthetics inhibit excitatory synapses and enhance inhibitory synapses in various preparations. The inhalational anesthetic isoflurane clearly can inhibit neurotransmitter release, while the small reduction in presynaptic action potential amplitude produced by isoflurane (3% reduction at MAC concentration) substantially inhibits neurotransmitter release. Inhalational anesthetics also can act postsynaptically, altering the response to released neurotransmitter. These actions are thought to be due to specific interactions of anesthetic agents with neurotransmitter receptors.
Intravenous anesthetics produce a narrower range of physiological effects. Their predominant actions are at the synapse, where they have profound and relatively specific effects on the postsynaptic response to released neurotransmitter. Most of the IV agents act predominantly by enhancing inhibitory neurotransmission, whereas ketamine predominantly inhibits excitatory neurotransmission at glutamatergic synapses.
MOLECULAR ACTIONS OF GENERAL ANESTHETICS. Most IV general anesthetics act predominantly through GABAA receptors and perhaps through some interactions with other ligand-gated ion channels such as NMDA receptors and 2-pore K+ channels.
Chloride channels gated by the inhibitory GABAA receptors (see Figures 14–3 and 14–6) are sensitive to a wide variety of anesthetics, including the halogenated inhalational agents and many IV agents (propofol, barbiturates, etomidate, and neurosteroids). At clinical concentrations, general anesthetics increase the sensitivity of the GABAA receptor to GABA, thus enhancing inhibitory neurotransmission and depressing nervous system activity. The action of anesthetics on the GABAA receptor probably is mediated by binding of the anesthetics to specific sites on the GABAA-receptor protein (but they do not compete with GABA for its binding site on the receptor). The capacity of propofol and etomidate to inhibit the response to noxious stimuli is mediated by a specific site on the β3 subunit of the GABAA receptor, whereas the sedative effects of these anesthetics are mediated by on the β2 subunit.
Structurally related to the GABAA receptors are other ligand-gated ion channels including glycine receptors and neuronal nicotinic ACh receptors. Glycine receptors may play a role in mediating inhibition by anesthetics of responses to noxious stimuli. Inhalational anesthetics enhance the capacity of glycine to activate glycine-gated chloride channels (glycine receptors), which play an important role in inhibitory neurotransmission in the spinal cord and brainstem. Propofol, neurosteroids, and barbiturates also potentiate glycine-activated currents, whereas etomidate and ketamine do not. Subanesthetic concentrations of the inhalational anesthetics inhibit some classes of neuronal nicotinic ACh receptors, which seem to mediate other components of anesthesia such as analgesia or amnesia.
Ketamine, nitrous oxide, cyclopropane, and xenon are the only general anesthetics that do not have significant effects on GABAA or glycine receptors. These agents inhibit a different type of ligand-gated ion channel, the NMDA receptor (see Figure 14–7 and Table 14–1). NMDA receptors are glutamate-gated cation channels that are somewhat selective for Ca++ and are involved in long-term modulation of synaptic responses (long-term potentiation) and glutamate-mediated neurotoxicity.
Halogenated inhalational anesthetics activate some members of a class of K+ channels known as 2-pore domain channels; other 2-pore domain channel family members are activated by xenon, nitrous oxide, and cyclopropane. These channels are located in both presynaptic and postsynaptic sites. The postsynaptic channels may be the molecular locus through which these agents hyperpolarize neurons.
Anatomic Sites of Anesthetic Action. In principle, general anesthetics could interrupt nervous system function at myriad levels, including peripheral sensory neurons, the spinal cord, the brainstem, and the cerebral cortex. Most anesthetics cause, with some exceptions, a global reduction in cerebral metabolic rate (CMR) and in cerebral blood flow (CBF). A consistent feature of general anesthesia is a suppression of metabolism in the thalamus, which serves as a major relay by which sensory input from the periphery ascends to the cortex. Suppression of thalamic activity may serve as a switch between the awake and anesthetized states. General anesthesia also results in the suppression of activity in specific regions of the cortex, including the mesial parietal cortex, posterior cingulate cortex, precuneus, and inferior parietal cortex.
Similarities between natural sleep and the anesthetized state suggest that anesthetics might also modulate endogenous sleep regulating pathways, which include ventrolateral preoptic (VLPO) and tuberomammillary nuclei. VLPO projects inhibitory GABA-ergic fibers to ascending arousal nuclei, which in turn project to the cortex, forebrain, and subcortical areas; release of histamine, 5HT, orexin, NE, and ACh mediate wakefulness. Intravenous and inhalational agents with activity at GABAA receptors can increase the inhibitory effects of VLPO, thereby suppressing consciousness. Dexmedetomidine, an α2 agonist, also increases VLPO-mediated inhibition by suppressing the inhibitory effect of locus ceruleus neurons on VLPO. Finally, both IV and inhalational anesthetics depress hippocampal neurotransmission, a probable locus for their amnestic effects.
PARENTERAL ANESTHETICS
Parenteral anesthetics are the most common drugs used for anesthetic induction of adults. Their lipophilicity, coupled with the relatively high perfusion of the brain and spinal cord, results in rapid onset and short duration after a single bolus dose. These drugs ultimately accumulate in fatty tissue. Each anesthetic has its own unique set of properties and side effects (Tables 19–1 and 19–2). Propofol and thiopental are the 2 most commonly used parenteral agents. Propofol is advantageous for procedures where rapid return to a preoperative mental status is desirable. Thiopental has a long-established track record of safety. Etomidate usually is reserved for patients at risk for hypotension and/or myocardial ischemia. Ketamine is best suited for patients with asthma or for children undergoing short, painful procedures.
Table 19–1
Pharmacological Properties of Parenteral Anesthetics
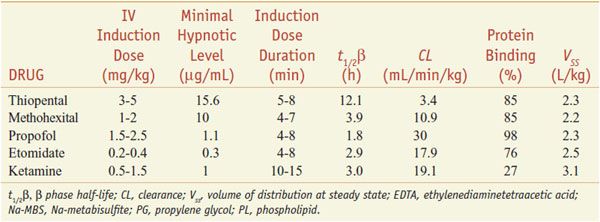
Table 19–2
Some Pharmacological Effects of Parenteral Anestheticsa
PHARMACOKINETIC PRINCIPLES
Parenteral anesthetics are small, hydrophobic, substituted aromatic or heterocyclic compounds (Figure 19–1). Hydrophobicity is the key factor governing their pharmacokinetics. After a single IV bolus, these drugs preferentially partition into the highly perfused and lipophilic tissues of the brain and spinal cord where they produce anesthesia within a single circulation time. Subsequently blood levels fall rapidly, resulting in drug redistribution out of the CNS back into the blood. The anesthetic then diffuses into less perfused tissues such as muscle and viscera, and at a slower rate into the poorly perfused but very hydrophobic adipose tissue. Termination of anesthesia after single boluses of parenteral anesthetics primarily reflects redistribution out of the CNS rather than metabolism (Figure 19–2).
Figure 19–1 Structures of some parenteral anesthetics.
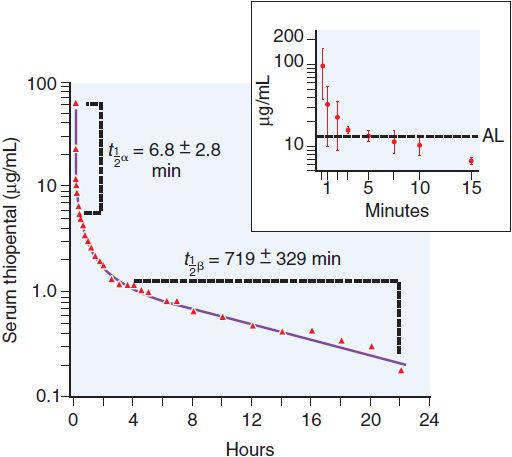
Figure 19–2 Thiopental serum levels after a single intravenous induction dose. Thiopental serum levels after a bolus can be described by two time constants, t1/2 α and t1/2 β. The initial fall is rapid (t1/2α<10 min) and is due to redistribution of drug from the plasma and the highly perfused brain and spinal cord into less well-perfused tissues such as muscle and fat. During this redistribution phase, serum thiopental concentration falls to levels at which patients awaken (AL, awakening level; see inset—the average thiopental serum concentration in 12 patients after a 6-mg/kg intravenous bolus of thiopental). Subsequent metabolism and elimination is much slower and is characterized by a half-life (t1/2 β) of more than 10 hours. (Adapted with permission from Burch PG, and Stanski DR, The role of metabolism and protein binding in thiopental anesthesia. Anesthesiology, 1983, 58:146–152. Copyright Lippincott Williams & Wilkins. http://lww.com.)
After redistribution, anesthetic blood levels fall according to a complex interaction between the metabolic rate and the amount and lipophilicity of the drug stored in the peripheral compartments. Thus, parenteral anesthetic half-life are “context-sensitive,” and the degree to which a t1/2 is contextual varies greatly from drug to drug, as might be predicted based on their differing hydrophobicities and metabolic clearances (Figure 19–3; see Table 19–1). For example, after a single bolus of thiopental, patients usually emerge from anesthesia within 10 min; however, a patient may require more than a day to awaken from a prolonged thiopental infusion. Most individual variability in sensitivity to parenteral anesthetics can be accounted for by pharmacokinetic factors. For example, in patients with lower cardiac output, the relative perfusion of the brain and the fraction of anesthetic dose delivered to the brain are higher; thus, patients in septic shock or with cardiomyopathy usually require lower doses of anesthetic. The elderly also typically require a smaller anesthetic dose, primarily because of a smaller initial volume of distribution.
Table 19–3
Properties of Inhalational Anesthetic Agents
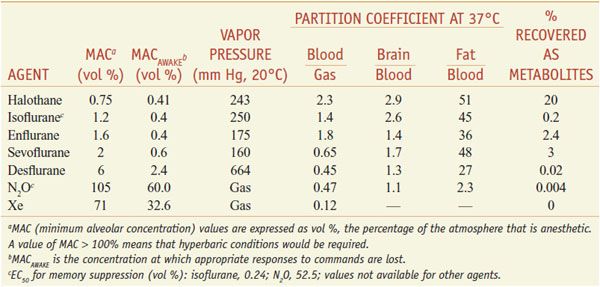
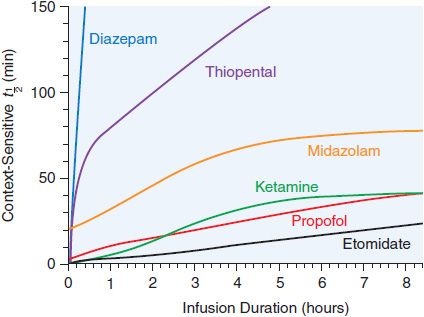
Figure 19–3 Context-sensitive half-time of general anesthetics. The duration of action of single intravenous doses of anesthetic/hypnotic drugs is similarly short for all and is determined by redistribution of the drugs away from their active sites (see Figure 19–2). However, after prolonged infusions, drug half-lives and durations of action are dependent on a complex interaction between the rate of redistribution of the drug, the amount of drug accumulated in fat, and the drug’s metabolic rate. This phenomenon has been termed the context-sensitive half-time; that is, the t1/2 of a drug can be estimated only if one knows the context—the total dose and over what time period it has been given. Note that the half-times of some drugs such as etomidate, propofol, and ketamine increase only modestly with prolonged infusions; others (e.g., diazepam and thiopental) increase dramatically. (Reproduced with permission from Reves JG, Glass PSA, Lubarsky DA, et al: Intravenous anesthetics, in Miller RD, et al, (eds): Miller’s Anesthesia, 7th ed. Philadelphia: Churchill Livingstone, 2010, p 718. Copyright © Elsevier.)
SPECIFIC PARENTERAL AGENTS
PROPOFOL, FOSPROPOFOL
Propofol is the most commonly used parenteral anesthetic in the U.S. Fospropofol is a prodrug form that is converted to propofol in vivo. The clinical pharmacological properties of propofol are summarized in Table 19–1.
CHEMISTRY AND FORMULATIONS. The active ingredient in propofol, 2,6-diisopropylphenol, is an oil at room temperature and insoluble in aqueous solutions. Propofol is formulated for IV administration as a 1% (10 mg/mL) emulsion in 10% soybean oil, 2.25% glycerol, and 1.2% purified egg phosphatide. In the U.S., disodium EDTA (0.05 mg/mL) or sodium metabisulfite (0.25 mg/mL) is added to inhibit bacterial growth. Propofol should be administered within 4 h of its removal from sterile packaging; unused drug should be discarded. The lipid emulsion formulation of propofol is associated with significant pain on injection and hyperlipidemia. A new aqueous formulation of propofol, fospropofol, which is not associated with these adverse effects, has recently been approved for use for sedation in patients undergoing diagnostic procedures. Fospropofol, which itself is inactive, is a phosphate ester prodrug of propofol that is hydrolyzed by endothelial alkaline phosphatases to yield propofol, phosphate, and formaldehyde. The formaldehyde is rapidly converted to formic acid, which then is metabolized by tetrahydrofolate dehydrogenase to CO2 and water.
DOSAGE AND CLINICAL USE. The induction dose of propofol (DIPRIVAN, others) in a healthy adult is 2-2.5 mg/kg. Dosages should be reduced in the elderly and in the presence of other sedatives and increased in young children. Because of its reasonably short elimination t1/2, propofol often is used for maintenance of anesthesia as well as for induction. For short procedures, small boluses (10-50% of the induction dose) every 5 min or as needed are effective. An infusion of propofol produces a more stable drug level (100-300 µg/kg/min) and is better suited for longer-term anesthetic maintenance. Sedating doses of propofol are 20-50% of those required for general anesthesia. Fospropofol produces dose-dependent sedation and can be administered in otherwise healthy individuals at 2-8 mg/kg intravenously (delivered either as a bolus or by a short infusion over 5-10 min). The optimum dose for sedation is ~6.5 mg/kg. This results in a loss of consciousness in ∼10 min. The duration of the sedative effect is ∼45 min.
PHARMACOKINETICS AND METABOLISM. Onset and duration of anesthesia after a single bolus are similar to thiopental. Propofol has a context-sensitive t1/2 of 10 min with an infusion lasting 3 h and ∼40 min for infusions lasting up to 8 h (see Figure 19–3). Propofol’s shorter duration of action after infusion can be explained by its very high clearance, coupled with the slow diffusion of drug from the peripheral to the central compartment.
Propofol is metabolized in the liver by conjugation to sulfate and glucuronide to less active metabolites that are renally excreted. Propofol is highly protein bound, and its pharmacokinetics, like those of the barbiturates, may be affected by conditions that alter serum protein levels. Clearance of propofol is reduced in the elderly. In neonates, propofol clearance is also reduced. By contrast, in young children, a more rapid clearance in combination with a larger central volume may necessitate larger doses of propofol for induction and maintenance of anesthesia.
The t1/2 for hydrolysis of fospropofol is 8 min; the drug has a small volume of distribution and a terminal t1/2 ∼46 min.
SIDE EFFECTS
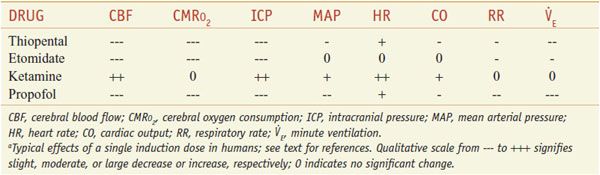
Nervous System. The sedation and hypnotic actions of propofol are mediated by its action on GABAA receptors; agonism at these receptors results in an increased chloride conduction and hyperpolarization of neurons. Propofol suppresses the EEG, and in sufficient doses, can produce burst suppression of the EEG. Propofol decreases cerebral metabolic rate of O2 consumption (CMRO2), CBF, and intracranial and intraocular pressures by about the same amount as thiopental. Like thiopental, propofol has been used in patients at risk for cerebral ischemia; however, no human outcome studies have been performed to determine its efficacy as a neuroprotectant.
Cardiovascular System. Propofol produces a dose-dependent decrease in blood pressure that is significantly greater than that produced by thiopental. The fall in blood pressure can be explained by both vasodilation and possibly mild depression of myocardial contractility. Propofol appears to blunt the baroreceptor reflex and reduce sympathetic nerve activity. As with thiopental, propofol should be used with caution in patients at risk for or intolerant of decreases in blood pressure.
Respiratory System. Propofol produces a slightly greater degree of respiratory depression than thiopental. Patients given propofol should be monitored to ensure adequate oxygenation and ventilation. Propofol appears to be less likely than barbiturates to provoke bronchospasm and may be the induction agent of choice in asthmatics. The bronchodilator properties of propofol may be attenuated by the metabisulfite preservative in some propofol formulations.
Other Side Effects. Propofol has a significant antiemetic action. Propofol elicits pain on injection that can be reduced with lidocaine and the use of larger arm and antecubital veins. A rare but potentially fatal complication, termed propofol infusion syndrome (PRIS), has been described primarily in prolonged, higher-dose infusions of propofol in young or head-injured patients. The syndrome is characterized by metabolic acidosis, hyperlipidemia, rhabdomyolysis, and an enlarged liver. The side-effect profile of fospropofol is similar to that of propofol.
ETOMIDATE
Etomidate is a substituted imidazole that is supplied as the active d-isomer. Etomidate is poorly soluble in water and is formulated as a 2 mg/mL solution in 35% propylene glycol. Unlike thiopental, etomidate does not induce precipitation of neuromuscular blockers or other drugs frequently given during anesthetic induction.
DOSAGE AND CLINICAL USE. Etomidate (AMIDATE, others) is primarily used for anesthetic induction of patients at risk for hypotension. Induction doses of etomidate (see Table 19–1) are accompanied by a high incidence of pain on injection and myoclonic movements. Lidocaine effectively reduces the pain of injection, while myoclonic movements can be reduced by premedication with either benzodiazepines or opiates. Etomidate is pharmacokinetically suitable for off-label infusion for anesthetic maintenance (10 μg/kg/min) or sedation (5 μg/kg/min); however, long-term infusions are not recommended.
PHARMACOKINETICS AND METABOLISM. An induction dose of etomidate has a rapid onset; redistribution limits the duration of action. Metabolism occurs in the liver, primarily to inactive compounds. Elimination is both renal (78%) and biliary (22%). Compared to thiopental, the duration of action of etomidate increases less with repeated doses (see Figure 19–3).
SIDE EFFECTS
Nervous System. Etomidate produces hypnosis and has no analgesic effects. The effects of etomidate on CBF, metabolism, and intracranial and intraocular pressures are similar to those of thiopental (without dropping mean arterial blood pressure). Etomidate produces increased EEG activity in epileptogenic foci and has been associated with seizures.
Cardiovascular System. Cardiovascular stability after induction is a major advantage of etomidate over either barbiturates or propofol. Induction doses of etomidate typically produce a small increase in heart rate and little or no decrease in blood pressure or cardiac output etomidate has little effect on coronary perfusion pressure while reducing myocardial O2 consumption.
Respiratory and Other Side Effects. The degree of respiratory depression due to etomidate appears to be less than that due to thiopental. Like methohexital, etomidate may induce hiccups but does not significantly stimulate histamine release. Etomidate has been associated with nausea and vomiting. The drug also inhibits adrenal biosynthetic enzymes required for the production of cortisol and some other steroids. Thus, while etomidate is not recommended for long-term infusion, it appears safe for anesthetic induction and has some unique advantages in patients prone to hemodynamic instability.
KETAMINE
Ketamine is an arylcyclohexylamine, a congener of phencyclidine. Ketamine is supplied as a mixture of the R+ and S-isomers even though the S-isomer is more potent with fewer side effects. Although more lipophilic than thiopental, ketamine is water-soluble.
DOSAGE AND CLINICAL USE. Ketamine (KETALAR, others) it useful for anesthetizing patients at risk for hypotension and bronchospasm and for certain pediatric procedures. However, significant side effects limit its routine use. Ketamine rapidly produces a hypnotic state quite distinct from that of other anesthetics. Patients have profound analgesia, unresponsiveness to commands, and amnesia, but may have their eyes open, move their limbs involuntarily, and breathe spontaneously. This cataleptic state has been termed dissociative anesthesia. The administration of ketamine has been shown to reduce the development of tolerance to long-term opioid use. Ketamine typically is administered intravenously but also is effective by intramuscular, oral, and rectal routes. Ketamine does not elicit pain on injection or true excitatory behavior as described for methohexital, although involuntary movements produced by ketamine can be mistaken for anesthetic excitement.
PHARMACOKINETICS AND METABOLISM. The onset and duration of an induction dose of ketamine are determined by the same distribution/redistribution mechanisms operant for all the other parenteral anesthetics. Ketamine is hepatically metabolized to norketamine, which has reduced CNS activity; norketamine is further metabolized and excreted in urine and bile. Ketamine has a large volume of distribution and rapid clearance that make it suitable for continuous infusion without the lengthening in duration of action seen with thiopental (see Table 19–1 and Figure 19–3). Protein binding is much lower with ketamine than with the other parenteral anesthetics.
SIDE EFFECTS
Nervous System. Ketamine has indirect sympathomimetic activity and produces distinct behavioral effects. The ketamine-induced cataleptic state is accompanied by nystagmus with pupillary dilation, salivation, lacrimation, and spontaneous limb movements with increased overall muscle tone. Patients are amnestic and unresponsive to painful stimuli. Ketamine produces profound analgesia, a distinct advantage over other parenteral anesthetics. Unlike other parenteral anesthetics, ketamine increases CBF and intracranial pressure (ICP) with minimal alteration of cerebral metabolism. The effects of ketamine on CBF can be readily attenuated by the simultaneous administration of sedative-hypnotic agents.
Emergence delirium, characterized by hallucinations, vivid dreams, and delusions, is a frequent complication of ketamine that can result in serious patient dissatisfaction and can complicate postoperative management. Benzodiazepines reduce the incidence of emergence delirium.
Cardiovascular System. Unlike other anesthetics, induction doses of ketamine typically increase blood pressure, heart rate, and cardiac output. The cardiovascular effects are indirect and are most likely mediated by inhibition of both central and peripheral catecholamine reuptake. Ketamine has direct negative inotropic and vasodilating activity, but these effects usually are overwhelmed by the indirect sympathomimetic action. Thus, ketamine is a useful drug, along with etomidate, for patients at risk for hypotension during anesthesia. While not arrhythmogenic, ketamine increases myocardial O2 consumption and is not an ideal drug for patients at risk for myocardial ischemia.
Respiratory System. The respiratory effects of ketamine are perhaps the best indication for its use. Induction doses of ketamine produce small and transient decreases in minute ventilation, but respiratory depression is less severe than with other general anesthetics. Ketamine is a potent bronchodilator and is particularly well suited for anesthetizing patients at high risk for bronchospasm.
BARBITURATES
CHEMISTRY AND FORMULATIONS. Barbiturates are derivatives of barbituric acid with either an oxygen or a sulfur at the 2-position (see Figure 19–1 and Chapter 17). The 3 barbiturates most commonly used in clinical anesthesia are sodium thiopental, thiamylal, and methohexital. Sodium thiopental (PENTOTHAL, others) has been used most frequently for inducing anesthesia. Thiamylal (SURITAL) is licensed in the U.S. only for veterinary use. Barbiturates are supplied as racemic mixtures despite enantioselectivity in their anesthetic potency. Barbiturates are formulated as the sodium salts with 6% sodium carbonate and reconstituted in water or isotonic saline to produce 2.5% (thiopental), 2% (thiamylal), or 1% (methohexital) alkaline solutions (10 ≤ pH ≤ 11). Mixing barbiturates with drugs in acidic solutions during anesthetic induction can result in precipitation of the barbiturate as the free acid; thus, standard practice is to delay the administration of other drugs until the barbiturate has cleared the IV tubing.
PHARMACOLOGICAL PROPERTIES. The pharmacological properties and other therapeutic uses of the barbiturates are presented in Chapter 17. Table 17–2 lists the common barbiturates with their clinical pharmacological properties.
DOSAGES AND CLINICAL USE. Recommended IV dosing for parenteral barbiturates in a healthy young adult is given in Table 19–1.
PHARMACOKINETICS AND METABOLISM. The principal mechanism limiting anesthetic duration after single doses is redistribution of these hydrophobic drugs from the brain to other tissues. However, after multiple doses or infusions, the duration of action of the barbiturates varies considerably depending on their clearances. See Table 19–1 for pharmacokinetic parameters.
Methohexital differs from the other two IV barbiturates in its much more rapid clearance; thus, it accumulates less during prolonged infusions. Because of their slow elimination and large volumes of distribution, prolonged infusions or very large doses of thiopental and thiamylal can produce unconsciousness lasting several days. All 3 barbiturates are primarily eliminated by hepatic metabolism and renal excretion of inactive metabolites; a small fraction of thiopental undergoes desulfuration to the longer-acting hypnotic pentobarbital. These drugs are highly protein bound. Hepatic disease or other conditions that reduce serum protein concentration will increase the initial free concentration and hypnotic effect of an induction dose.
SIDE EFFECTS
Nervous System. Barbiturates suppress the EEG and can produce burst suppression of the EEG. They reduce the CMR, as measured by CMRO2, in a dose-dependent manner. As a consequence of the decrease in CMRO2, CBF and ICP are similarly reduced. Presumably in part due to their CNS depressant activity, barbiturates are effective anticonvulsants. Thiopental in particular is a proven medication in the treatment of status epilepticus. Methohexital can increase ictal activity, and seizures have been described in patients who received doses sufficient to produce burst suppression of the EEG. This property makes methohexital a good choice for anesthesia in patients who undergo electroconvulsive therapy.
Cardiovascular System. The anesthetic barbiturates produce dose-dependent decreases in blood pressure. The effect is due primarily to vasodilation, particularly venodilation, and to a lesser degree to a direct decrease in cardiac contractility. Typically, heart rate increases as a compensatory response to a lower blood pressure, although barbiturates also blunt the baroreceptor reflex. Thiopental maintains the ratio of myocardial O2 supply to demand in patients with coronary artery disease within a normal blood pressure range. Hypotension can be severe in patients with an impaired ability to compensate for venodilation such as those with hypovolemia, cardiomyopathy, valvular heart disease, coronary artery disease, cardiac tamponade, or β adrenergic blockade. None of the barbiturates has been shown to be arrhythmogenic.
Respiratory System.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


