CHAPTER 43 G2 Phase and Control of Entry into Mitosis
The G2 phase was originally defined simply as a gap between the completion of DNA replication and the onset of mitosis. In fact, definition of this phase is not entirely straightforward, as progression from interphase into mitosis is gradual, with early mitotic prophase sharing characteristics of both interphase and mitosis. Here, we define the G2 phase as the period from the end of the S phase until mid-prophase, that is, until activation of the main mitotic kinase, Cdk1–cyclin B1.
Enzymology of the G2/Mitosis Transition
The transition between the G2 phase and mitosis is the most profound morphologic and physiological change that occurs during the life of a growing cell, matched only by the dramatic changes that occur during death by apoptosis (see Chapter 46). Entry into mitosis is controlled by a network of stimulatory and inhibitory protein kinases and phosphatases, presided over by Cdk1–cyclin B1. (Chapter 40 introduced the components involved in the G2/M transition.)
Cdk1, the driving force for entry into mitosis, is present at a constant level throughout the cell cycle. Cdk1 regulation is multifaceted (see Fig. 40-14), including binding of cyclin cofactors, inhibition and activation by phosphorylation, binding of inhibitory molecules, and changes in subcellular localization.
Mammalian cells have at least three B-type cyclins: B1, B2, and B3. Cyclin B1 is essential for triggering the G2/M transition, and disruption of its gene has lethal consequences. Cyclin B1, newly synthesized during the latter part of the cell cycle, binds Cdk1 and shuttles it in and out of the nucleus. Importin b carries the Cdk1–cyclin B1 complex into the nucleus, and then Crm1 rapidly exports it back to the cytoplasm (see Chapter 16). Cdk1–cyclin B2 associates with the Golgi apparatus during interphase and might function in Golgi disassembly during mitosis (see Fig. 44-4). Cyclin B3 appears to function only during meiosis in mammals.
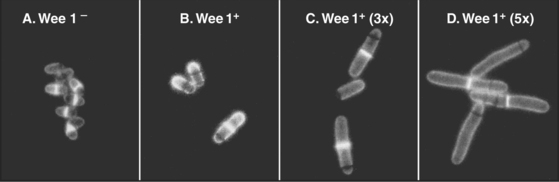
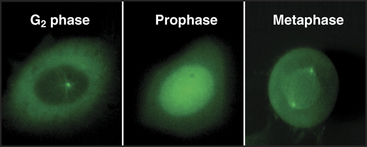
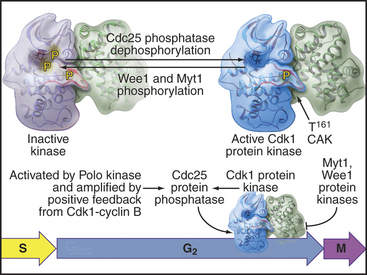
As cells approach the G2/M transition, a combination of one stimulatory and two inhibitory kinases control activation of Cdk1–cyclin B1. The activating kinase, called CAK (Cdk-activating kinase; actually Cdk7–cyclin H), is located in the nucleus. CAK phosphorylates Cdk1 on T161, allowing the refolding of the active site cleft that is required for the enzyme to bind substrates (see Chapter 40). Two other kinases, Wee1 and Myt1, counteract the action of CAK (Fig. 43-1). Wee1 is localized to the nucleus, where it inhibits Cdk1 by phosphoryla-ting Y15 adjacent to the ATP-binding site. The role of Wee1 as a mitotic inhibitor is clearly demonstrated in S. pombe: Overexpression of functional Wee1 delays or prevents the entry of cells into mitosis (Fig. 43-2). Myt1 associated with the Golgi apparatus and endoplasmic reticulum inhibits Cdk1 while it resides in the cytoplasm by phosphorylating T14 and Y15. Together, Wee1 and Myt1 ensure that Cdk1 remains inactive as it shuttles into and out of the nucleus (Fig. 43-3). This combination of stimulatory and inhibitory modifica-tions holds Cdk1–cyclin B1 poised for a burst of activation.
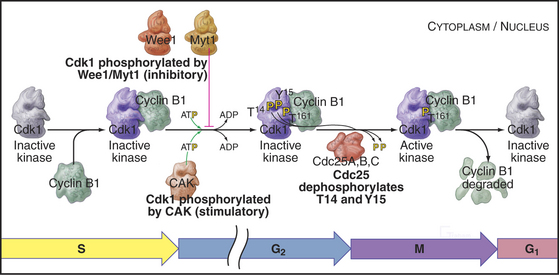
Figure 43-1 Regulation of Cdk1 by cyclin binding and protein phosphorylation from the late S phase through mid-mitosis.
(Structure for Wee1 kinase provided by E. N. Baker prior to re-lease to the Protein Data Bank; based on Squire CJ, Dickson JM, Ivanovic I, Baker EN: Structure of human Wee1A kinase: Kinase domain complexed with inhibitor PD0407824. Structure 13:541–550, 2005. PDB file: 1X8B.)
The three Cdc25 protein phosphatases remove the inhibitory phosphates from Cdk1–cyclin B1 and trigger the G2/M transition. Cdc25s are dual-specificity protein phosphatases (see Fig. 25-5) that remove phosphates from serine (S), threonine (T), and tyrosine (Y) residues, including the inhibitory phosphates from T14 and Y15 of Cdk1. Cdc25s are regulated by stimulatory and inhibitory phosphorylation, by alterations in their subcellular localization, and by ubiquitin-mediated proteolysis. Cdc25A, the only one of the three to be indispensable for life, functions at both the G1/S and G2/M transitions, whereas Cdc25B and Cdc25C have roles in the G2/M transition. The preferred targets of the individual Cdc25 isoforms are not known.
Cdc25A and Cdc25C are relatively inactive during interphase for three reasons (Fig. 43-4). First, phosphorylation on a serine residue creates a binding site for a member of the 14-3-3 group of adapter proteins (see Fig. 25-10). These proteins bind sites on target proteins containing serines flanked by several other characteristic amino acids, but only when the critical serine is phosphorylated. This is an example of the general mechanism whereby phosphorylation regu-lates interactions between proteins in response to physiological signals (see Chapter 25). Association of 14-3-3 with Cdc25A interferes with its binding to Cdk1–cyclin B1 and inhibits its import into the nucleus. Because Cdc25C has an intrinsic nuclear export sequence, the 14-3-3-bound form of Cdc25C is primarily cytoplasmic.
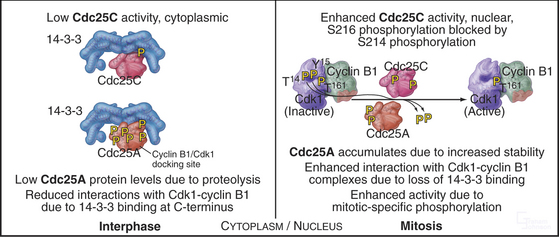
Figure 43-4 Regulation of Cdc25A and Cdc25C activity in interphase and mitosis.
(Based on an original figure by Helen Piwnica-Worms.)
Changes in Subcellular Localization at the G2/M Transition
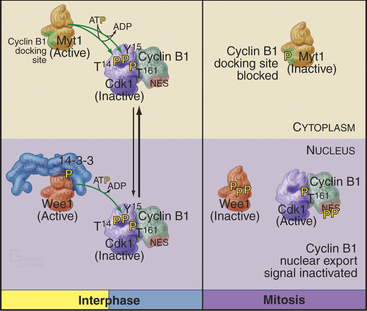
Cyclin B1 (with associated Cdk1) and Cdc25C have both nuclear import and nuclear export signals, so they shuttle in and out of the nucleus throughout interphase (Fig. 43-5). Cdc25A resides in the nucleus. During interphase, Cdk1–cyclin B1 and Cdc25C spend most of their time in the cytoplasm. Inhibitory phosphorylation of Cdk1 and Cdc25C keeps Cdk1 activity low.
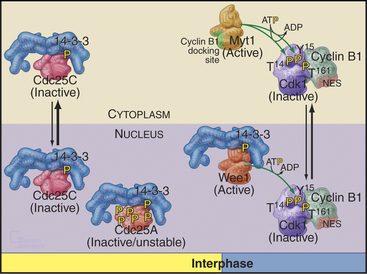
Early in prophase, phosphorylation of cyclin B1 inactivates its nuclear export signal and promotes its nuclear import (see Chapter 16), allowing Cdk1–cyclin B1 to accumulate rapidly in the nucleus within five minutes (Figs. 43-3 and 43-6). Cdc25C also stops shuttling at the G2/M transition, probably as a result of phosphorylation by Polo kinase. The best evidence seems to indicate that the Cdk1–cyclin B that accumulates in the nucleus is already active, but restriction of Cdk–cyclin B complexes to the nucleus together with Cdc25A and Cdc25C significantly increases their local concentration (the volume of the nucleus is much less than the volume of the cytoplasm) and might contribute to the final burst of Cdk1–cyclin B1 activation.
Cdk1–Cyclin A and the Initiation of Prophase
As is noted in Chapter 42, Cdk2–cyclin A plays a critical role during the S phase. Several lines of evidence have revealed that a Cdk–cyclin A complex also helps to trigger the G2/M transition. First, inactivation of cyclin A, either by mutation in Drosophila or by injection of anti–cyclin A antibodies into cultured cells, arrests the cell cycle in G2. Second, Cdk–cyclin A activity peaks at G2/M, before the peak of Cdk1–cyclin B1 activity. Finally, if activated Cdk2–cyclin A complexes are injected into cells just after completion of the S phase, cells enter mitosis prematurely.
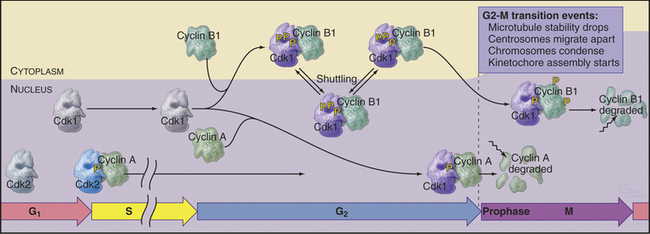
Cdk–cyclin A kinase is most likely to regulate several events at the transition from the G2 phase to prophase, including changes in microtubule behavior and chromosome condensation. Late in G2, the half-life of microtubules drops dramatically from about 10 minutes to about 30 seconds (see Table 44-1). This, coupled with centrosomes’ enhanced ability to initiate microtubule polymerization, completely transforms the organization of the microtubule cytoskeleton. Centrosomes take on the appearance of spindle poles and migrate apart over the surface of the nucleus. At the same time, chromatin begins to condense in the nucleus. The mechanism of chromosome condensation is not known, but a protein complex called condensin is required for this condensation to begin during prophase (see Fig. 13-19). Condensin does not associate tightly with chromosomes during interphase; association with chromosomes requires phosphorylation of two of its subunits by a Cdk. This occurs at G2/M. As chromosomes condense, several mitosis-specific kinetochore proteins also move into the nucleus and associate with kinetochores (see Chapter 13).
These events occur while most Cdk1–cyclin B1 is in the cytoplasm. It is most likely, therefore, that Cdk1–cyclin A triggers at least the nuclear events of prophase (Fig. 43-7). In fact, microinjection of a specific inhibitor of Cdk1–cyclin A causes prophase cells to return rapidly to interphase; chromosomes decondense, rounded prophase cells flatten, and the interphase microtubule network returns. Commitment to mitosis appears to be irreversible only after Cdk1–cyclin B1 enters the nucleus.
Summary of the Main Events of the G2/M Transition
Synthesis of cyclin B1 in the latter portion of the S and G2 phases leads to assembly of Cdk1–cyclin B heterodimers that shuttle into and out of the nucleus, spending most of their time in the cytoplasm associated with microtubules. In the late S and G2 phases, activation of Cdk1–cyclin A initiates mitotic prophase, beginning with changes in microtubule dynamics and chromosome condensation. Several events trigger entry into the active phase of mitosis. Cdc25A becomes stabilized and no longer binds 14-3-3 proteins. This results in the accumulation of Cdc25A and allows more efficient interactions between Cdc25A and Cdk1–cyclin B. The phosphate-binding site for 14-3-3 proteins on Cdc25C becomes dephosphorylated, allowing it to accumulate in the nucleus. In addition, phosphorylation of cyclin B1 blocks its export from the nucleus and promotes its import, thus causing Cdk1–cyclin B1 to accumulate rapidly in the nucleus. The inhibitory kinase Wee1 is also dephosphorylated, and its activity drops. Cdc25A and Cdc25C activate Cdk1–cyclin B1 by removing inhibitory phosphates on T14 and Y15. This starts in the cytoplasm and then may be stimulated as the proteins concentrate in the nucleus. There, the action of Cdk1–cyclin B1 on the nuclear lamina triggers nuclear envelope breakdown and drives the cell into mitosis. Fig. 43-8 summarizes the network regulating Cdk1–cyclin B activation.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


