Extrarenal Rhabdoid Tumor
Cyril Fisher, MD, DSc, FRCPath
Key Facts
Terminology
Malignant neoplasm of polygonal cells with characteristic large cytoplasmic inclusion and eccentric large nucleus
Requires exclusion of specific tumor types with rhabdoid cytomorphology
Can be component of other tumors as composite rhabdoid tumor
Clinical Issues
Majority in childhood, including congenitally
Deep soft tissue or skin
Visceral locations
Aggressive with frequent local recurrence and metastasis
Prognosis very poor
Microscopic Pathology
Polygonal cells in discohesive sheets
Ancillary Tests
Most cases express cytokeratin or epithelial membrane antigen
Absence of immunoreactivity for INI1 is diagnostically useful
CD34(-)
Chromosome 22q11.2 deletions
Ultrastructurally, large cytoplasmic inclusion comprising whorl of intermediate filaments 8-10 microns in diameter
Diagnostic Checklist
Exclude specific tumor subtype with rhabdoid cell morphology
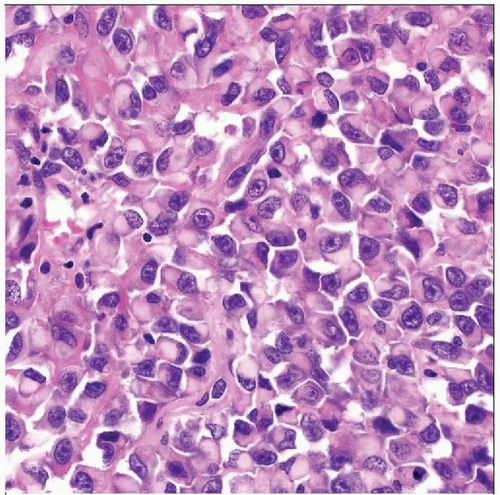 Hematoxylin & eosin shows sheets of rhabdoid cells without architecture. The cells have amphophilic cytoplasm and eccentric rounded nuclei with large nucleoli and are often discohesive, as seen here. |
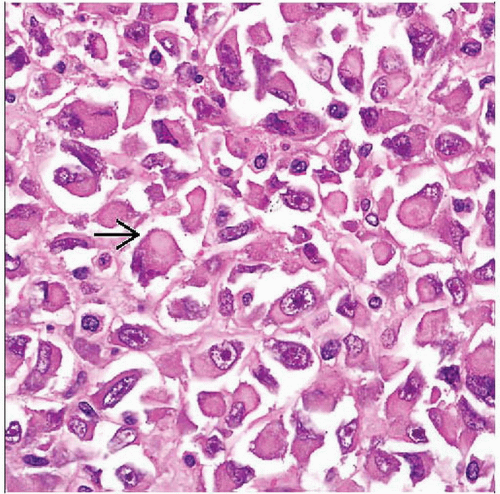 Hematoxylin & eosin shows cell detail, with abundant cytoplasm containing pale-staining inclusions  . These are composed of cytokeratin- and vimentin-intermediate filaments. Note the prominent nucleoli. . These are composed of cytokeratin- and vimentin-intermediate filaments. Note the prominent nucleoli. |
TERMINOLOGY
Abbreviations
Extrarenal rhabdoid tumor (ERT)
Synonyms
Malignant rhabdoid tumor
Atypical teratoid/rhabdoid tumor: Term for similar neoplasm in central nervous system
Definitions
Rare malignant neoplasm of characteristic polygonal cells
Large nuclei with prominent nucleoli
Abundant eosinophilic cytoplasm, which displaces nucleus to 1 side
Requires exclusion of specific tumor types with occasional rhabdoid cytomorphology
Extraskeletal myxoid chondrosarcoma
Leiomyosarcoma
Myoepithelial tumor
Gastrointestinal stromal tumor
Endometrial stromal sarcoma
Synovial sarcoma
Mesothelioma
Carcinoma
Melanoma
Can be pure or form part of specific tumor type (composite ERT)
ETIOLOGY/PATHOGENESIS
Genetic Factors
Some have abnormalities of chromosome 22q11.2
Some patients have germline mutations of hSNF5/SMARCB1 gene
Some associated with myofibroma- or hamartoma-like cutaneous lesions
Rarely multiple
CLINICAL ISSUES
Epidemiology
Incidence
Rare
Age
Majority in childhood, including congenitally
Rare examples in adults after all mimics excluded
Site
Deep soft tissue or skin
Axial and paraxial, cervical or paravertebral regions, vulva, perineum
Thigh, limb girdles
Viscera
GI tract, liver, heart, bladder, brain
Presentation
Rapidly growing mass, occasionally ulcerates
Natural History
Aggressive with frequent local recurrence and metastasis
Treatment
Surgical approaches
Excision where feasible
Drugs
Chemotherapy
Rarely effective
Prognosis
Very poor
MACROSCOPIC FEATURES
General Features
Multinodular, nonencapsulated, poorly circumscribed
Pale or tan, with hemorrhage and necrosis
Size
Up to 5 cm or more at presentation
MICROSCOPIC PATHOLOGY
Histologic Features



