Estrogens and Progestins
Estrogens and progestins are hormones that produce myriad physiological actions. In women, these include developmental effects, neuroendocrine actions involved in the control of ovulation, the cyclical preparation of the reproductive tract for fertilization and implantation, and major actions on mineral, carbohydrate, protein, and lipid metabolism. Estrogens also have important actions in males, including effects on bone, spermatogenesis, and behavior. The most common uses of these agents are menopausal hormone therapy and contraception in women, but the specific compounds and dosages used in these 2 settings differ substantially. Anti-estrogens are used in the treatment of hormone-responsive breast cancer and infertility. Selective estrogen receptor modulators (SERMs) that display tissue-selective agonist or antagonist activities are useful to prevent breast cancer and osteoporosis. The main use of antiprogestins has been for medical abortion.
ESTROGENS
Estrogens interact with 2 receptors of the nuclear receptor superfamily, termed estrogen receptors α (Erα) and β (ERβ). The most potent naturally occurring estrogen in humans, for both ERα- and ERβ-mediated actions, is 17β-estradiol, followed by estrone and estriol. Steroidal estrogens arise from androstenedione or testosterone (Figure 40–1) by a reaction catalyzed by aromatase (CYP19).
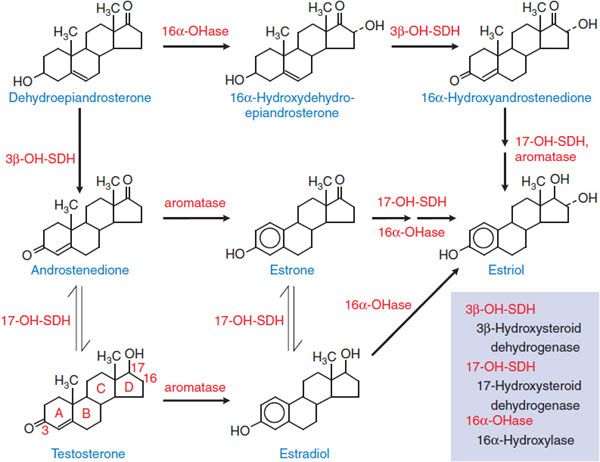
Figure 40–1 The biosynthetic pathway for estrogens.
The ovaries are the principal source of circulating estrogen in premenopausal women, with estradiol the main secretory product. Gonadotropins, acting via receptors that couple to the Gs-adenylyl cyclase–cyclic AMP pathway, increase the activities of aromatase and facilitate the transport of cholesterol (the precursor of all steroids) into the mitochondria of cells that synthesize steroids. In the ovary, type I 17β-hydroxysteroid dehydrogenase favors the production of testosterone and estradiol from androstenedione and estrone, respectively. In the liver, the type II enzyme favors oxidation of circulating estradiol to estrone, and both of these steroids are then converted to estriol (see Figure 40–1). All 3 of these estrogens are excreted in the urine along with their glucuronide and sulfate conjugates.
In postmenopausal women, the principal source of circulating estrogen is adipose tissue stroma, where estrone is synthesized from dehydroepiandrosterone secreted by the adrenals. In men, estrogens are produced by the testes, but extragonadal production by aromatization of circulating C19 steroids (e.g., androstenedione and dehydroepiandrosterone) accounts for most circulating estrogens. Local production of estrogens by aromatization of androgens may play a causal or promotional role in the development of certain diseases such as breast cancer. Estrogens also may be produced from androgens via aromatase in the central nervous system (CNS) and other tissues and exert local effects near their production site (e.g., in bone they affect bone mineral density).
Human urine during pregnancy is an abundant source of natural estrogens. Pregnant mare’s urine is the source of conjugated equine estrogens, which have been widely used therapeutically for many years.
PHYSIOLOGICAL AND PHARMACOLOGICAL ACTIONS
DEVELOPMENTAL ACTIONS. Estrogens are largely responsible for pubertal changes in girls and secondary sexual characteristics.
Estrogens cause growth and development of the vagina, uterus, and fallopian tubes, and contribute to breast enlargement. They also contribute to molding the body contours, shaping the skeleton, and causing the pubertal growth spurt of the long bones and epiphyseal closure. Growth of axillary and pubic hair, pigmentation of the genital region, and the regional pigmentation of the nipples and areolae that occur after the first trimester of pregnancy are also estrogenic actions. Androgens may also play a secondary role in female sexual development (see Chapter 41). Estrogens also play important developmental roles in males. In boys, estrogen deficiency diminishes the pubertal growth spurt and delays skeletal maturation and epiphyseal closure so that linear growth continues into adulthood. Estrogen deficiency in men leads to elevated gonadotropins, macroorchidism, and increased testosterone levels and also may affect carbohydrate and lipid metabolism and fertility in some individuals.
NEUROENDOCRINE CONTROL OF THE MENSTRUAL CYCLE. A neuroendocrine cascade involving the hypothalamus, pituitary, and ovaries controls the menstrual cycle (Figure 40–2). A neuronal oscillator, or “clock,” in the hypothalamus fires at intervals that coincide with the release of gonadotropin-releasing hormone (GnRH) into the hypothalamic-pituitary portal vasculature (see Chapter 38). GnRH interacts with its receptor on pituitary gonadotropes to cause release of luteinizing hormone (LH) and follicle-stimulating hormone (FSH). The frequency of the GnRH pulses, which varies in the different phases of the menstrual cycle, controls the relative synthesis of FSH and LH.
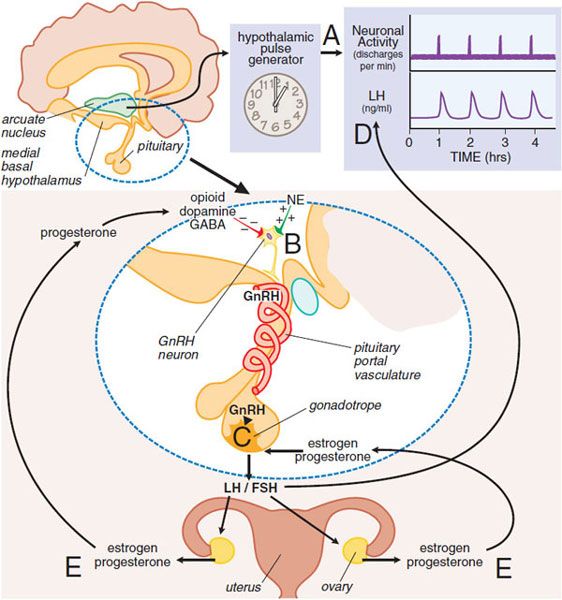
Figure 40–2 Neuroendocrine control of gonadotropin secretion in females. The hypothalamic pulse generator located in the arcuate nucleus of the hypothalamus functions as a neuronal “clock” that fires at regular hourly intervals (A). This results in the periodic release of gonadotropin-releasing hormone (GnRH) from GnRH-containing neurons into the hypothalamic-pituitary portal vasculature (B). GnRH neurons (B) receive inhibitory input from opioid, dopamine, and GABA neurons and stimulatory input from noradrenergic neurons (NE, norepinephrine). The pulses of GnRH trigger the intermittent release of luteinizing hormone (LH) and follicle-stimulating hormone (FSH) from pituitary gonadotropes (C), resulting in the pulsatile plasma profile (D). FSH and LH regulate ovarian production of estrogen and progesterone, which exert feedback controls (E). (See text and Figure 40–3 for additional details.)
The gonadotropins (LH and FSH) regulate the growth and maturation of follicles in the ovary and the ovarian production of estrogen and progesterone, which exert feedback regulation on the pituitary and hypothalamus. Because the release of GnRH is intermittent, secretion of LH and FSH is pulsatile. The pulse frequency is determined by the neural “clock” (see Figure 40–2), termed the hypothalamic GnRH pulse generator, but the amount of gonadotropin released in each pulse (i.e., the pulse amplitude) is largely controlled by the actions of estrogens and progesterone on the pituitary. The intermittent, pulsatile nature of hormone release is essential for the maintenance of normal ovulatory menstrual cycles because constant infusion of GnRH results in a cessation of gonadotropin release and ovarian steroid production (see Chapter 38). Although the precise mechanism that regulates the timing of GnRH release (i.e., pulse frequency) is unclear, hypothalamic cells appear to have an intrinsic ability to release GnRH episodically. Ovarian steroids, primarily progesterone, regulate the frequency of GnRH release. At puberty the pulse generator is activated and establishes cyclic profiles of pituitary and ovarian hormones. Although the mechanism of activation is not entirely established, it may involve increases in circulating insulin-like growth factor 1 (IGF)-1 and leptin levels, the latter acting to inhibit neuropeptide Y (NPY) in the arcuate nucleus to relieve an inhibitory effect on GnRH neurons.
Figure 40–3 provides a schematic diagram of the profiles of gonadotropin and gonadal steroid levels in the menstrual cycle. In the early follicular phase of the cycle: (1) the pulse generator produces bursts of neuronal activity with a frequency of about 1/h that correspond with pulses of GnRH secretion, (2) these cause a corresponding pulsatile release of LH and FSH from pituitary gonadotropes, and (3) FSH in particular causes the graafian follicle to mature and secrete estrogen. The effects of estrogens on the pituitary are inhibitory at this time and cause the amount of LH and FSH released from the pituitary to decline, so gonadotropin levels gradually fall. Estrogens act primarily on the pituitary to control the amplitude of gonadotropin pulses and may also contribute to the amplitude of GnRH pulses secreted by the hypothalamus. Inhibin, produced by the ovary, exerts negative feedback to selectively decrease serum FSH.
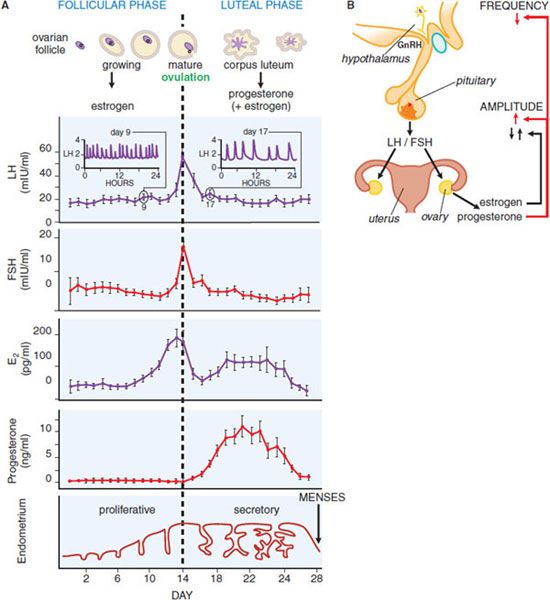
Figure 40–3 Hormonal relationships of the human menstrual cycle. A. Average daily values of LH, FSH, estradiol (E2), and progesterone in plasma samples from women exhibiting normal 28-day menstrual cycles. Changes in the ovarian follicle (top) and endometrium (bottom) also are illustrated schematically. Frequent plasma sampling reveals pulsatile patterns of gonadotropin release. Characteristic profiles are illustrated schematically for the follicular phase (day 9, inset on left) and luteal phase (day 17, inset on right). Both the frequency (number of pulses per hour) and amplitude (extent of change of hormone release) of pulses vary throughout the cycle. (Redrawn with permission from Thorneycroft et al., 1971. © Elsevier). B. Major regulatory effects of ovarian steroids on hypothalamic-pituitary function. Estrogen decreases the amount of follicle-stimulating hormone (FSH) and luteinizing hormone (LH) released (i.e., gonadotropin pulse amplitude) during most of the cycle and triggers a surge of LH release only at mid-cycle. Progesterone decreases the frequency of GnRH release from the hypothalamus and thus decreases the frequency of plasma gonadotropin pulses. Progesterone also increases the amount of LH released (i.e., the pulse amplitude) during the luteal phase of the cycle.
At mid-cycle, serum estradiol rises above a threshold level of 150-200 pg/mL for ~36 h, exerting a brief positive feedback effect on the pituitary to trigger the preovulatory surge of LH and FSH. This effect primarily involves a change in pituitary responsiveness to GnRH. Progesterone may contribute to the mid-cycle LH surge. The mid-cycle surge in gonadotropins stimulates follicular rupture and ovulation within 1-2 days. The ruptured follicle then develops into the corpus luteum, which produces large amounts of progesterone and lesser amounts of estrogen under the influence of LH during the second half of the cycle. In the absence of pregnancy, the corpus luteum ceases to function, steroid levels drop, and menstruation occurs. When steroid levels drop, the pulse generator reverts to a firing pattern, the entire system then resets, and a new ovarian cycle occurs.
In the follicular phase of the cycle, estrogens inhibit gonadotropin release but then have a brief mid-cycle stimulatory action that increases the amount released and causes the LH surge. Progesterone, acting on the hypothalamus, exerts the predominant control of the frequency of LH release. It decreases the firing rate of the hypothalamic pulse generator, an action thought to be mediated largely via inhibitory opioid neurons (containing progesterone receptors) that synapse with GnRH neurons. Progesterone also exerts a direct effect on the pituitary to oppose the inhibitory actions of estrogens and thus enhance the amount of LH released (i.e., to increase the amplitude of the LH pulses). These steroid feedback effects, coupled with the intrinsic activity of the hypothalamic GnRH pulse generator, lead to relatively frequent LH pulses of small amplitude in the follicular phase of the cycle, and less frequent pulses of larger amplitude in the luteal phase.
In males, testosterone regulates the hypothalamic-pituitary-gonadal axis at both the hypothalamic and pituitary levels, and its negative feedback effect is mediated to a substantial degree by estrogen formed via aromatization.
EFFECTS OF CYCLICAL GONADAL STEROIDS ON THE REPRODUCTIVE TRACT
The cyclical changes in estrogen and progesterone production by the ovaries regulate corresponding events in the fallopian tubes, uterus, cervix, and vagina. Physiologically, these changes prepare the uterus for implantation, and the proper timing of events in these tissues is essential for pregnancy. If pregnancy does not occur, the endometrium is shed as the menstrual discharge (see Figure 40–3).
Menstruation marks the start of the menstrual cycle. During the follicular (or proliferative) phase of the cycle, estrogen begins the rebuilding of the endometrium by stimulating proliferation and differentiation. An important response to estrogen in the endometrium and other tissues is induction of the progesterone receptor (PR), which enables cells to respond to this hormone during the second half of the cycle. In the luteal (or secretory) phase of the cycle, elevated progesterone limits the proliferative effect of estrogens on the endometrium by stimulating differentiation. Major effects include stimulation of epithelial secretions important for implantation of the blastocyst and the characteristic growth of the endometrial blood vessels seen at this time. These effects are mediated by PR-A in animal models. Progesterone is thus important in preparation for implantation and for the changes that take place in the uterus at the implantation site (i.e., the decidual response). There is a narrow “window of implantation,” spanning days 19-24 of the endometrial cycle, when the epithelial cells of the endometrium are receptive to blastocyst implantation. If implantation occurs, human chorionic gonadotropin (hCG), produced initially by the trophoblast and later by the placenta, interacts with the LH receptor of the corpus luteum to maintain steroid hormone synthesis during the early stages of pregnancy. Later the placenta becomes the major site of estrogen and progesterone synthesis.
METABOLIC EFFECTS. Estrogens affect myriad tissues. Many nonreproductive tissues, including bone, vascular endothelium, liver, CNS, immune system, the GI tract, and heart, express low levels of both estrogen receptors (ERs), and the ratio of ERe to ERβ varies in a cell-specific manner. The effects of estrogens on selected aspects of mineral, lipid, carbohydrate, and protein metabolism are particularly important for understanding their pharmacological actions.
Long-term administration of estrogen is associated with decreased plasma renin, angiotensin-converting enzyme, and endothelin-1; expression of the AT1 receptor for AngII is also decreased. Estrogen actions on the vascular wall include increased production of NO and prostacyclin. All of these changes promote vasodilation and retard atherogenesis. Estrogens alter a number of metabolic pathways that affect the clotting cascade. Systemic effects include changes in hepatic production of plasma proteins. Estrogens cause a small increase in coagulation factors II, VII, IX, X, and XII, and they decrease the anticoagulation factors protein C, protein S, and antithrombin III (see Chapter 30). Fibrinolytic pathways also are affected, and several studies of women treated with estrogen alone or estrogen with a progestin have demonstrated decreased levels of plasminogen-activator inhibitor 1 (PAI-1) protein with a concomitant increase in fibrinolysis. Thus, estrogens increase both coagulation and fibrinolytic pathways, and imbalance in these 2 opposing activities may cause adverse effects.
Estrogens increase bone mass. Bone is continuously remodeled by the resorptive action of osteoclasts and the bone-forming action of osteoblasts. Estrogens directly regulate osteoblasts and increase osteocyte survival by inhibiting apoptosis. The major effect of estrogens is to decrease the number and activity of osteoclasts.
Estrogens slightly elevate serum triglycerides and slightly reduce total serum cholesterol levels. They increase high-density lipoprotein (HDL) levels and decrease the levels of low-density lipoprotein (LDL) and lipoprotein A (LPA) (see Chapter 31). The presence of ERs in the liver suggests that the beneficial effects of estrogen on lipoprotein metabolism may be due partly to direct hepatic actions. Estrogens also alter bile composition by increasing cholesterol secretion and decreasing bile acid secretion. This leads to increased saturation of bile with cholesterol and appears to be the basis for increased gallstone formation in some women receiving estrogens. In general, estrogens increase plasma levels of cortisol-binding globulin, thyroxine-binding globulin, and sex hormone-binding globulin (SHBG), which binds both androgens and estrogens.
ESTROGEN RECEPTORS
Estrogens exert their effects by interaction with receptors that are members of the superfamily of nuclear receptors. The 2 estrogen receptor genes are located on separate chromosomes: ESR1 encodes ERα, and ESR2 encodes ERβ. Both ERs are estrogen-dependent nuclear transcription factors that have different tissue distributions and transcriptional regulatory effects on a wide number of target genes. Both ERα and ERβ exist as multiple mRNA isoforms due to differential promoter use and alternative splicing. There are significant differences between the 2 receptor isoforms in the ligand-binding domains and in both transactivation domains. Human ERβ does not appear to contain a functional AF-1 domain. The receptors appear to have different biological functions and respond differently to various estrogenic compounds. However, their high homology in the DNA-binding domains suggests that both receptors recognize similar DNA sequences and hence regulate many of the same target genes.
ERα is expressed most abundantly in the female reproductive tract—especially the uterus, vagina, and ovaries—as well as in the mammary gland, the hypothalamus, endothelial cells, and vascular smooth muscle. ERβ is expressed most highly in the prostate and ovaries, with lower expression in lung, brain, bone, and vasculature. Both forms of ER are expressed on breast cancers, although ERβ is believed to be the predominant form responsible for growth regulation (see Chapter 63). Polymorphic variants of ER have been identified, but attempts to correlate specific polymorphisms with the frequency of breast cancer, bone mass, endometrial cancer, or cardiovascular disease have led to contradictory results.
A cloned G protein-coupled receptor, GPR30, also appears to interact with estrogens in some cell systems, and its participation in the rapid effects of estrogen is an attractive idea. There may be interaction/cross-talk between membrane-associated ERα and membrane-localized GPR30 in some cancer cells, but in vivo confirmation is lacking.
MECHANISM OF ACTION
Both ERs are ligand-activated transcription factors that increase or decrease the transcription of target genes (Figure 40–4). After entering the cell by passive diffusion through the plasma membrane, the hormone binds to an ER in the nucleus. In the nucleus, the ER is present as an inactive monomer bound to heat-shock protein 90 (HSP90), and upon binding estrogen, a change in ER conformation dissociates the heat-shock proteins and causes receptor dimerization, which increases the affinity and the rate of receptor binding to DNA. Homodimers of ERα or ERβ and ERα/ERβ heterodimers can be produced depending on the receptor complement in a given cell. The ER dimer binds to estrogen response elements (EREs), typically located in the promoter region of target genes. The ER/DNA complex recruits a cascade of coactivator and other proteins to the promoter region of target genes (Figure 40–4B and legend) and allows the proteins that make up the general transcription apparatus to assemble and initiate transcription.
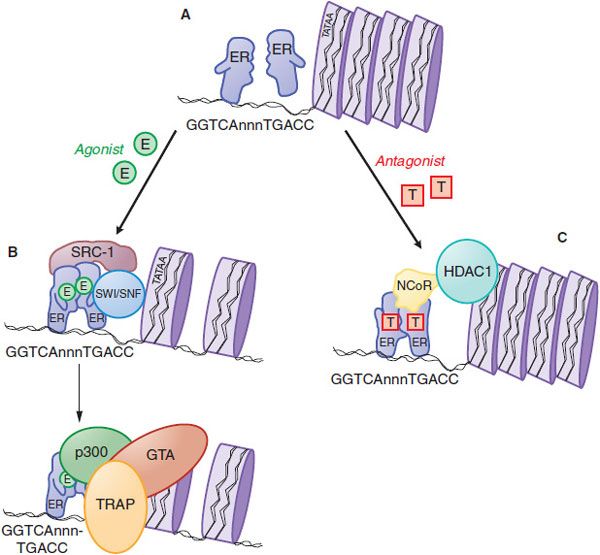
Figure 40–4 Molecular mechanism of action of nuclear estrogen receptor. A. Unliganded estrogen receptor (ER) exists as a monomer within the nucleus. B. Agonists such as 17β-estradiol (E) bind to the ER and cause a ligand-directed change in conformation that facilitates dimerization and interaction with specific estrogen response elements in DNA. The ER-DNA complex recruits coactivators such as SWI/SNF that modify chromatin structure, and coactivators such as steroid-receptor coactivator-1 (SRC-1) that has histone acetyltransferase (HAT) activity that further alters chromatin structure. This remodeling facilitates the exchange of the recruited proteins such that other coactivators (e.g., p300 and the TRAP complex) associate on the target gene promoter and proteins that comprise the general transcription apparatus (GTA) are recruited, with subsequent synthesis of mRNA. C. Antagonists such as tamoxifen (T) also bind to the ER but produce a different receptor conformation. The antagonist-induced conformation also facilitates dimerization and interaction with DNA, but a different set of proteins called corepressors, such as nuclear-hormone receptor corepressor (NcoR), are recruited to the complex. NcoR further recruits proteins such as histone deacetylase I (HDAC1) that act on histones to stabilize nucleosome structure and prevent interaction with the GTA.
Some ERs are located on the plasma membrane of cells. These ERs are encoded by the same genes that encode ERα and ERβ but are transported to the plasma membrane and reside mainly in caveolae. Translocation to the membrane by all sex steroid receptors is mediated by palmitoylation of a 9–amino acid motif common to these receptors. Membrane-localized ERs mediate the rapid activation of some proteins such as MAPK (phosphorylated in several cell types) and the rapid increase in cyclic AMP caused by the hormone. These membrane interactions and sequelae provide additional levels of cross-talk and complexity in estrogen signaling.
ADME. Various estrogens are available for oral, parenteral, transdermal, or topical administration. Given the lipophilic nature of estrogens, absorption generally is good with the appropriate preparation. Aqueous or oil-based esters of estradiol are available for intramuscular injection, ranging in frequency from every week to once per month. Conjugated estrogens are available for IV or IM administration. Transdermal patches that are changed once or twice weekly deliver estradiol continuously through the skin. Preparations are available for topical use in the vagina or for application to the skin. Estrogen preparations also are available in combination with a progestin. All estrogens are labeled with precautionary statements urging the prescribing of the lowest effective dose and for the shortest duration consistent with the treatment goals and risks for each individual patient.
Oral administration is common and may use estradiol, conjugated estrogens, esters of estrone and other estrogens, and ethinyl estradiol (in combination with a progestin). Estradiol is available in nonmicronized (FEMTRACE) and micronized preparations (ESTRACE, others). The micronized formulations yield a large surface for rapid absorption to partially overcome low absolute oral bioavailability due to first-pass metabolism. Addition of the ethinyl substituent at C17 (ethinyl estradiol) inhibits first-pass hepatic metabolism. Other common oral preparations contain conjugated equine estrogens (PREMARIN), which are primarily the sulfate esters of estrone, equilin, and other naturally occurring compounds; esterified esters (MENEST); or mixtures of synthetic conjugated estrogens prepared from plant-derived sources (CENESTIN;ENJUVIA). These are hydrolyzed by enzymes present in the lower gut that remove the charged sulfate groups and allow absorption of estrogen across the intestinal epithelium. In another oral preparation, estropipate (ORTHO-EST, OGEN, others), estrone is solubilized as the sulfate and stabilized with piperazine. Due largely to differences in metabolism, the potencies of various oral preparations differ widely; e.g., ethinyl estradiol is much more potent than conjugated estrogens.
Administration of estradiol via transdermal patches (ALORA, CLIMARA, ESTRADERM, VIVELLE, others) provides slow, sustained release of the hormone, systemic distribution, and more constant blood levels than oral dosing. Estradiol is also available as a topical emulsion (ESTRASORB) or as a gel (ESTROGEL). The transdermal route does not lead to the high level of the drug in the portal circulation after oral administration, and thus should minimize hepatic effects of estrogens. Preparations available for intramuscular injection include compounds such as estradiol valerate (DELESTROGEN, others) or estradiol cypionate (DEPO-ESTRADIOL, others), which may be absorbed over several weeks following a single intramuscular injection. Preparations of estradiol (ESTRACE) and conjugated estrogen (PREMARIN) creams are available for topical administration to the vagina. A 3-month vaginal ring (ESTRING, FEMRING) may be used for slow release of estradiol, and tablets are also available for vaginal use (VAGIFEM).
Estradiol, ethinyl estradiol, and other estrogens are extensively bound to plasma proteins. Estradiol and other naturally occurring estrogens are bound mainly to SHBG; ethinyl estradiol is bound extensively to serum albumin but not SHBG. Due to their size and lipophilic nature, unbound estrogens distribute rapidly and extensively. Estrogens undergo rapid hepatic biotransformation, with a plasma t1/2 measured in minutes. Estrogens also undergo enterohepatic recirculation via (1) sulfate and glucuronide conjugation in the liver, (2) biliary secretion of the conjugates into the intestine, and (3) hydrolysis in the gut (largely by bacterial enzymes) followed by reabsorption. Estradiol is converted primarily by 17β-hydroxysteroid dehydrogenase to estrone, which undergoes conversion by 16α-hydroxylation and 17-keto reduction to estriol, the major urinary metabolite; myriad sulfate and glucuronide conjugates appear in the urine. Ethinyl estradiol is cleared much more slowly than estradiol due to decreased hepatic metabolism with t1/2 of 13-27 h. Mestranol, a component of some combination oral contraceptives, is the 3-methyl ether of ethinyl estradiol.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


