| USAN: | Esomeprazole |
| Brand Name: | Nexium (AstraZeneca) |
| Molecular Weight: | 713.13 |
| FDA Approval: | 2001 |
| Drug Class: | Anti-Ulcer |
| Indications: | Gastroesophageal Reflux Disease (GERD) |
| Mechanism of Action: | Proton Pump Inhibitor (H+/K+-ATPase Inhibitor) |
■ 1 History of Ulcer and Ulcer Drugs
Gastric acid, hydrochloric acid (HCl), is essential to our digestion. It helps break down proteins, fats, and starches in our food into nutrients such as amino acids and carbohydrates that our body can absorb. Alas, there can be too much of a good thing. Too much secretion of gastric acid for too long can cause heartburn or acid reflux, also known as gastroesophageal reflux disease (GERD). In addition, having too much gastric acid for too long will damage the mucosa of the stomach and may cause ulcer and even stomach cancers!
While the exact pathogenesis of ulcers is not known, theories abound. It is still a popular belief that ulcer is caused by stress. Thirty years ago, the discovery of Helicobacter pylori demonstrated that the bacterium contributes to gastritis and peptic ulcers.1
Regardless of the root of stomach ulcers, they are closely associated with excessive secretion of gastric acid, so that was what early ulcer drugs intended to treat. Dozens of antacids are on the market to neutralize excess HCl: For instance, Mylanta and Maalox contain Al(OH)3 and Mg(OH)2. Tums contains CaCO3. Alka-Seltzer contains NaHCO3 (baking soda).
However, antacids only treat the symptoms of excessive acid secretion, and are therefore only effective as a short-term treatment. A better therapy would treat the causation of excessive acid secretion. Histamine-2 receptor antagonists work by blocking the receptors that are responsible for excessive acid secretion: the histamine-2 receptors. As a consequence, they are more efficacious and have fewer adverse effects in comparison to conventional antacids.
1.1 Histamine-2 Receptor Antagonists
The fact that histamine stimulates gastric acid secretion in the stomach was first observed by Popielski in the 1920s.
Popielski studied under Pavlov, who won the Nobel Prize (for Physiology or Medicine) in 1904 for his studies in digestive system, but who is now better known for his classical dog-conditioning experiments. As an independent researcher at the University of Kraków, Popielski discovered that histamine was a stimulant of gastric glands, and acted directly without the involvement of vagal nerves.2 After injecting histamine subcutaneously into gastric fistulas in dogs, Popielski observed copious gastric acid secretion and extremely high acidity in the dogs’ stomachs.3 The experiment solidly established the association between histamine and gastric acid secretion.
Interestingly enough, antihistamines used to treat allergies did not have significant impact on gastric acid secretion. This observation led Black at SmithKline & French (SK&F) to speculate in 1964 that there were two types of histamine receptors, and that selective histamine H2 receptor antagonists would be effective in decreasing gastric acid secretion and treating peptic ulcer.4 The fruit of SK&F’s endeavor was cimetidine (Tagamet, 2), which revolutionized the treatment of peptic ulcers. However, cimetidine (2), the prototype for H2 receptor antagonists, had a trio of shortcomings: short half-life, DDI, and occasionally causing gynecomastia in some male patients due to its antiandrogenic activities. Ranitidine (Zantac, 3), famotidine (Pepcid, 4), and nizatidine (Axid, 5) are more potent and more selective and also safer than cimetidine (2). They (3–5) are devoid of the trio of the drawbacks associated with cimetidine (2).5–7



The H2 receptor antagonist approach produced many blockbuster drugs that revolutionized the pharmaceutical industry. Cimetidine (Tagamet, 2), in fact, was the first blockbuster drug, with annual sales over $1 billion.
1.2 Proton Pump Inhibitors
While H2 receptor antagonist approach were succeeding, other MOAs in treating ulcer were being pursued by many pharmaceutical companies. Hässle AB Sweden (now AstraZeneca) began their Gastrin Project in 1967. At the time, the rational drug design approach in drug discovery was not yet popular. Hässle tackled their Gastrin Project the old fashioned way: using animal models. Basically, they prepared compounds and tested them in fistula in dogs to see if the compound lowered the acidity of the dog’s stomach.8
Hässle first derived their inspirations from Servier’s CMN 131 (6), a thioamide and SK&F’s cimetidine (2) to arrive at H 77/67 (7) with an imidazoline moiety.9,10 Moving away from thioamide was understandable because of thioamide’s propensity to cause hepatotoxicity.11 Further development led to H 124/26 (8), which was unfortunately covered by a Hungarian patent. Fortuitously, its oxidative metabolite H 83/69 (timoprazole, 9) was even more active than sulfide 8. Unfortunately, timoprazole (9) was associated with thymus and thyroid toxicities. Hässle prepared picoprazole (10) in 1976.



Picoprazole (10) was later found to cause necrotizing vasculitis in the small intestine in some dogs when given too much for too long.12 Efforts were made to find safer analogs of picoprazole (10). Since 3,5-dimethyl-4-methoxy-pyridine was known to increase the basicity of a pyridine ring, Hässle prepares it without systematically explore the SAR. Omeprazole (11) tested as the most powerful inhibitor of stimulated gastric acid secretion in experimental animals at the time. The drug had no sign of serious toxicity in animal models.
Omeprazole (Prilosec, 11) was approved by the FDA in 1990 and was the world’s best-selling drug from 1996 to 2000, when it was unseated by atorvastatin calcium (Lipitor; see chapter 1).
Lansoprazole (Prevacid, 12) is another proton pump inhibitor (PPI) sold by TAP Pharmaceuticals, a joint venture of Abbott Laboratories and Takeda. Two other PPIs are pantoprazole (Protonix, 13) and rabeprazole (Aciphex, 14).13


In 1987, a group at Hässle embarked on a focused program to find a backup for Prilosec (11) with better bioavailability. Among all the compounds, only one was better than Prilosec: the S-(–)-diastereomer of omeprazole, esomeprazole (Nexium, 1). When tested in rats, Nexium was 4–5 times more bioavailable than the R-diastereomer (1‘).14 Another lucky break for Hässle was that they tested Nexium (1) directly in humans and saw similar effects to what they observed with rats. Later, when the two isomers were tested on dogs, no significant difference of efficacy was detected for the two isomers. In this case, rat was a better animal model than dog: Had they used dogs for their initial in vivo tests, they probably would never have found Nexium (1).15
Just as Prilosec’s patent was about to expire, AstraZeneca launched Nexium (1) in early 2001. The timing generated some controversy with regard to whether Prilosec (11) and Nexium (1) become the same molecule in the human body after metabolism. We will address the issue in the remainder of the chapter.
Nexium was the best-selling drug of the year in 2012 with annual sales of $6 billion. However, it lost patent protection in 2014.
■ 2 Pharmacology
2.1 Mechanism of Action
The MOA of Nexium (1) is through inhibition of an enzyme called gastric H+/K+-ATPase, also known as proton pump.
In the late 1970s, Sachs’s group showed that H+/K+-ATPase was the proton pump of the stomach.16 They also described immunologic detection of the pump in different organs using a polycolonal antibody against the ATPase. The enzyme reacted strongly with the stomach and weakly with the thyroid. In 1978, collaborating with Hässle, Sachs demonstrated that timoprazole (8) and picoprazole (9) were prodrugs but were converted to the active form after accumulation in the acidic secretory canaliculus of the parietal cells. “Thus arose an entirely new domain of peptic ulcer therapy,” claimed Modlin, a professor at Yale Medical School. 17
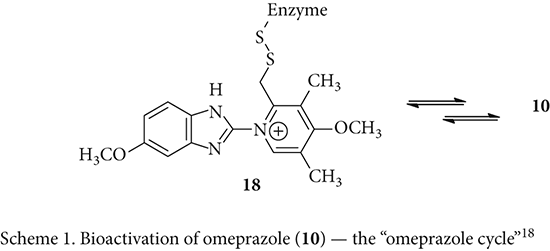
Similar to timoprazole (8) and picoprazole (9), omeprazole (10) itself is not active in inhibiting the ATPase in vitro. Rather, it is also a prodrug and becomes activated in vivo via the “omeprazole cycle” as shown in Scheme 1.18,19 Sequentially, protonation of 10 takes place slowly when it encounters the acidic medium in the stomach. The protonated benzimidazole intermediate 14 then undergoes an intramolecular nucleophilic cyclization in a 5-exo-dig fashion where the pyridine moiety serves as the nucleophile to furnish benzimidazoline 15. A reversible ring opening of 15 then delivers sulfenic acid 16, which serves as an electrophile in a 6-exo-trig ring-closure to afford the reactive sulfenamide intermediate 17 after dehydration. The sulfenamide 17 is a very good electrophile, and is readily attacked by the cysteine residue of the enzyme H+/K+-ATPase. Therefore, Prilosec (11) is a pro-drug and sulfenamide 17 is the actual inhibitory species.
Nexium (1) is the (S)-enantiomer of racemic Prilosec (11), which is a mixture of two enantiomers. The former has better pharmacokinetics and pharmacodynamics than the latter and therefore possesses higher efficacy in controlling acid secretion and has a better therapeutic profile (see below).



2.2 Structure–Activity Relationship
The SAR of Prilosec (11) was extensively investigated.14 An abridged SAR table is shown in Table 10.1. The inhibitory effect of some Prilosec analogs on a partially purified H+/K+-ATPase is represented as pIC50, which is the negative logarithm of its corresponding IC50 values. Meanwhile, pSA (SA stands for the quantity of sulfonamide, e.g., sulfenamide 17 in Fig. 10.1) is the negative logarithm of quantity (in mol/L of incubation mixture) of sulfenamide needed for 50% inhibition.20
From the earlier SAR, a generic structure of 19 possessing a substituted pyridine and a substituted benzimidazole was found to be the required core structure to have the antisecretory effect. When the Hässle team attempted to replace either pyridine or benzimidazole with other heterocycles, they observed no significant boost of the gastric acid inhibitory effect. Only a few close analogs of the benzimidazole, such as imidazole and aza-benzimidazole, showed weak inhibitory ability. This is readily explained using the “omeprazole cycle” because both pyridine and benzimidazole work in tandem to form the reactive sulfenamide intermediate such as 17.


The sulfur atom was ideal to be a sulfoxide. Both sulfide and sulfone were inactive in vitro. One-carbon linkers (–CH2S–, –CH2SO–, and –CH(CH3)SO–) all showed antisecretory activity in vivo. The fact that the sulfide is active in vivo is apparently due to its oxidation to the sulfoxide by CYP450. Elongating the linker between the sulfoxide and the pyridine did not offer any advantages, so one carbon linker was kept constant.
In entries 1–7, the pyridine rings with 3,5-(CH3)2, 4-CH3O substituents seem to be optimal. Because 3,5-dimethyl-4-methoxy-pyridine was known to increase the basicity of a pyridine ring, Hässle prepared it without systematically exploring the SAR. The result was that the pKa values of the pyridine ring was increased and the compounds containing the 3,5-dimethyl-4-methoxy-substituents were found to be most potent. This could also be explained by the omeprazole cycle. Since 3,5-dimethyl-4-methoxy-pyridine is the most basic, 14 donates electron pairs with the greatest ease, thus promoting the catalytic cycle. As luck would have it, later systemic SAR studies demonstrated that 3,5-dimethyl-4-methoxy-pyridine had higher in vivo potency than the corresponding 3-methyl-4-methoxy-pyridine and 5-methyl-4-methoxy-pyridine analogs.14
Substituents on the benzimidazole have a less profound impact on the potency of drugs. In entry 7, nitro compound 19f completely (100%) converted to the active sulfenamide (SA, similar to 17) within 30 min after dosing. It is possible that the nitro group is strongly electron-withdrawing, rendering the benzimidazole more prone to accept an electron pair from the pyridine ring, thus accelerating the catalytic cycle. Not surprisingly, another electron-withdrawing group methylsulfoxide showed similar acceleration of SA formation, albeit to a lesser extent. However, 19f was not chosen as a development candidate. It can be speculated that nitro-aromatics have been historically associated with liver toxicities so that a conscious decision was probably made to avoid it. In the end, 5-methoxy-substitution of benzimidazole (entry 2) was chosen not only because 10 is potent, but also based on other pharmaceutics considerations, such as stability, crystallinity, and the ease with which to handle the active pharmaceutical ingredient (API).
2.3 Bioavailability, Metabolism and Toxicology
Not surprisingly, the bioavailabilities of both Prilosec (11) and Nexium (1) are complicated by the fact that they are prodrugs and the original APIs are completely metabolized in vivo once they come in contact with gastric acid.21–26 This argues for the merit of using animal models rather than rational drug design, which may not find Prilosec (11) in the first place.
First-pass metabolism, sometimes also known as first-pass elimination, takes place largely in the liver where CYP450 enzymes oxidize the drug and make it more polar, and thus easier to eliminate. In clinical trials with young healthy volunteers, only 54% of a given dose of Prilosec (11) remained after dosing due to extensive first-pass metabolism.21 This means that within a couple of hours dosing, nearly half of the API has been absorbed and eliminated.22
Three major metabolism pathways for Prilosec (11) are shown in Fig. 10.1.23–25 All three are oxidative processes. On the left of the figure, under the influence of S-mephenytoin hydroxylase (S-MPH) and CYP3A, the 5-methyl group on the pyridine ring of omeprazole (10) is oxidized to the corresponding pyridylmethyl alcohol (20), which could be potentially further oxidized to the corresponding acid (see Fig. 10.2). Meanwhile, O-demethylation of the 4-methoxyl group on the pyridine ring provides a metabolite as 4-hydroxypyridine, which is unstable in the physiological environment.




