INTRODUCTION
LEARNING OBJECTIVES
After studying this chapter you should be able to:
Construct a flow diagram outlining the different causes of erythrocytosis.
Develop a coherent diagnostic strategy for evaluating a patient with erythrocytosis.
Some patients come to medical attention because of an increase in red cell mass, manifested by hematocrit and hemoglobin levels that exceed the upper limit of normal for age and sex. The terms “erythrocytosis” and “polycythemia” are often used interchangeably. The former is preferred because it covers all conditions in which the red cell mass is increased. “Polycythemia” is a more restrictive term, implying not only increased hemoglobin and hematocrit levels but also leukocytosis and thrombocytosis.
In the initial evaluation of the patient, it is important to consider whether the hematocrit and hemoglobin levels might be falsely elevated because of a reduction in plasma volume. False or “pseudo” erythrocytosis may be due to dehydration or loss of gastrointestinal fluid from vomiting or diarrhea. In addition to these acute conditions, modest elevations of hemoglobin (Hb) and hematocrit (Hct) levels may be caused by chronic contraction in plasma volume, most often encountered in hypertensive, overweight, middle-aged males, a condition known as stress erythrocytosis or Gaisböck syndrome. However, when levels exceed two standard deviations above normal (Hb >17.5 g/dL, Hct >54% in males; Hb >16.0 g/dL, Hct >46% in females), it is likely that the patient has true erythrocytosis.
Many individuals with erythrocytosis are asymptomatic. However, as the red cell mass increases, they develop a ruddy or florid facial complexion. With further increases, the patient may develop central nervous system symptoms, such as forgetfulness, lethargy, or headache, and may have signs of excess cardiac preload, such as distended neck veins and hepatic congestion. More often, the clinical presentation is dominated by symptoms and signs related to the underlying cause rather than the erythrocytosis per se.
Establishing the cause of erythrocytosis can be a diagnostic challenge that requires the application of sound pathophysiologic principles. Table 12-1 presents an etiologic framework based on different mechanisms responsible for the expansion of the red cell mass. In primary erythrocytosis, the increase is due to autonomous proliferation of erythroid progenitors in the marrow, independent of external cues. In these patients, the abundant oxygen supply from the elevated hemoglobin concentration dampens expression of the erythropoietin (Epo) gene, and, as a result, plasma Epo levels are low. In contrast, secondary erythrocytosis is caused by an increased plasma erythropoietin level, which is either an appropriate physiologic response to hypoxia or a result of inappropriate autonomous up-regulation of Epo expression.
PRIMARY ERYTHROCYTOSIS
The most commonly encountered form of primary erythrocytosis is an acquired myeloproliferative disorder called polycythemia vera. These patients have a marked expansion of the red cell mass, owing to acquisition of a mutation in the tyrosine kinase JAK2, a signaling enzyme that plays a crucial role in hematopoietic cell differentiation. The remarkable ability of this mutation to trigger autonomous erythroid proliferation is depicted in Figure 12-1B. In addition to elevations in hemoglobin and hematocrit levels, patients with polycythemia vera usually have leukocytosis, thrombocytosis, and modest splenomegaly. For more information on the molecular pathogenesis, clinical presentation, course, diagnosis, and treatment of this disorder, see Chapter 20.
FIGURE 12-1
Erythropoietin (Epo)-dependent signaling. A) Normal erythropoiesis. When Epo binds to its dimeric receptor on erythroid progenitor cells, the two intracellular tails are pulled together, allowing phosphorylation of JAK2 kinase, which initiates the signal transduction cascade. A phosphatase (Pase), that binds to the C-terminal portion of the cytosolic domain of the Epo receptor, acts as a negative regulator, limiting the extent of signal transduction. B) Patients with polycythemia vera have acquired a mutant JAK2 kinase, which has enhanced activity sufficient to initiate and maintain signal transduction in the absence of Epo. C) Some individuals have familial erythrocytosis owing to the dominant inheritance of a C-terminal deletion of the Epo receptor. This truncated receptor cannot bind to the phosphatase and thus gives rise to autonomous, high-level signal transduction, signified by P*.
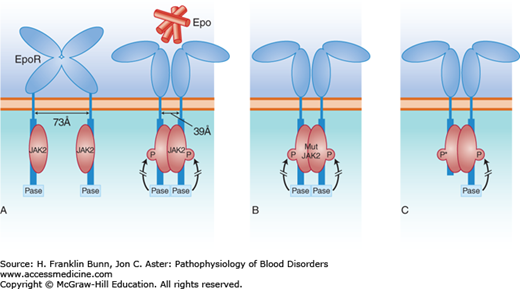
Rare families have been reported in which the polycythemia vera clinical phenotype is inherited in an autosomal dominant manner. The mutation(s) responsible for the familial disease are not yet known. Other families have autosomal dominant inheritance of autonomous erythrocytosis with normal white cell and platelet counts. One affected individual was an elite cross-country skier who won several world championships. He and a large number of relatives had hemoglobin levels as high as 19 g/dL, despite low plasma erythropoietin levels. DNA sequencing revealed that those having erythrocytosis were heterozygous for a stop codon mutation that caused the truncation of the intracellular tail of the Epo receptor. As depicted in Figure 12-1C, in the normal Epo receptor, a phosphatase, binds to this site and functions as a brake, limiting the potency of signaling by the JAK2 kinase. In the absence of this negative control, receptor-mediated signaling and erythropoiesis are markedly enhanced. Subsequently, a number of other families have been identified around the world with erythrocytosis due to several other mutations causing similar truncations in the Epo receptor.



