Erythrocytes
Carolyn Chiling Chai
Red blood cells (RBCs) decrease in size as they mature and lose the nucleus. The cytoplasm turns from blue to pink-orange as they acquire hemoglobin (Hb). The average diameter of a mature RBC is 7 to 8 μm and the average volume is 90 femtoliter (fL). The RBC membrane contains proteins including cytoskeleton and cell antigens. The main function of the RBC is producing hemoglobin.
Any RBC with a central pallor >3 μm is hypochromic. Any defect in hemoglobin synthesis leads to hypochromasia and decreased mean corpuscular hemoglobin concentration (MCHC). A typical example is iron-deficiency anemia. Hypochromasia may also be seen in lead poisoning and water artifact, although the central pallor in the latter is distinctly outlined. A hypochromic cell may not be microcytic (e.g., target cells). Conversely, normocytes or macrocytes may also be hypochromic. When RBCs are prematurely released to the circulation, they are large and polychromatic (blue-gray) due to residual cytoplasmic RNA and are called reticulocytes. The normal value of the reticulocytes is 0.5% to 2.5%. During RBC regeneration (effective erythropoiesis), as in hemorrhage, hemolysis, or treatment of anemia, the reticulocyte count and polychromasia are increased proportionately. Reticulocytes can be viewed with a supravital stain.
The main hematopoietic sites in adults are vertebrae, pelvis, ribs, sternum, skull, and the proximal ends of the femur and humer. There, the RBCs originate from a pleuripotent stem cell, the colony-forming unit-stem (CFU-S). During differentiation, it becomes erythroidcommitted burst-forming unit-erythroid (BFU-E), which matures into colony-forming unit-erythroid (CFU-E). Erythropoietin stimulates BFU-E and CFU-E to differentiate to pronormoblasts, the first precursor recognizable by light microscope, which gives rise to 16 mature erythrocytes through four cell divisions in 72 hours. These four divisions, in sequential order, produce basophilic normoblasts, polychromatophilic normoblasts, orthochromatophilic normoblasts, and reticulocytes. Reticulocytes eventually extrude RNA to become erythrocytes. Effective erythropoiesis represents the rate of release of newly formed RBCs from the bone marrow and ineffective erythropoiesis represents the rate of loss of potential erythrocytes as a result of phagocytosis (1, 2, 3, 4, 5, 6).
Most RBCs are destroyed in the reticuloendothelial system. The extravascular hemolysis indicates RBC destruction by phagocytosis in the reticuloendothelial system. The spleen is the principal site of erythrocyte phagocytosis with normal aging. The liver plays a more active role in the removal of damaged RBCs. Extremely damaged cells may lyse within the circulation before reaching the liver or the spleen; this is referred to as intravascular hemolysis.
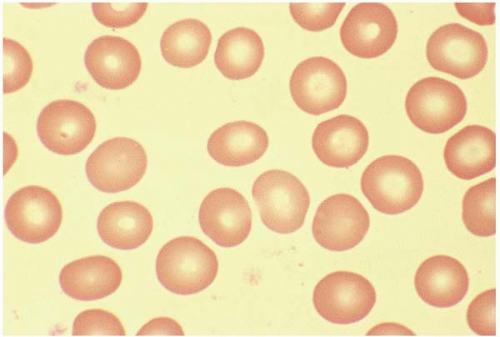 Figure 5.1 Normal RBC (normocytes) in the peripheral blood. |
OVERVIEW OF ERYTHROCYTE MORPHOLOGY
Red Cell Size
A normal RBC is a biconcaved disc with a diameter of 7 to 8 μm. A normocyte indicates an RBC with a mean cell volume (MCV) of 80 to 100 fL (Fig. 5.1).
Microcytes are RBCs with an MCV <80 fL. Any defect in Hb synthesis results in hypochromic microcytes (Fig. 5.2). The defect may be due to inadequate iron supply, iron absorption, iron release or iron utilization as in iron-deficiency anemia; or due to abnormal globin synthesis as in thalassemia and hemoglobinopathy. Although porphyrin synthesis is important for hemoglobin synthesis, its deficiency typically does not cause decreased RBC size.
Macrocytes are RBCs with an MCV >100 fL. Macrocyte formation may result from impaired DNA synthesis (megaloblastic erythropoiesis); accelerated erythropoiesis with early release of reticulocytes; or increased membrane cholesterol or lecithin as in liver disease. Macro-ovalocytes lack a central pallor and are characteristic of megaloblastic anemia (Fig. 5.3). Round macrocytes are seen in liver disease, chronic alcoholism, chemotherapy, hypothyroidism, postsplenectomy, and neonatal blood. In patients with mycoplasma pneumoniae infection or cold agglutinin disease, the MCV may be artificially elevated due to RBC aggregates.
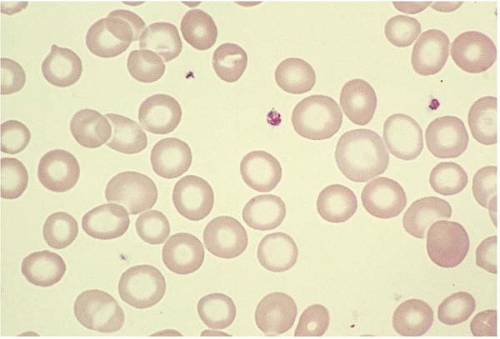 Figure 5.2 Hypochromic microcytic RBC in the peripheral blood smear from a patient with iron-deficiency anemia. |
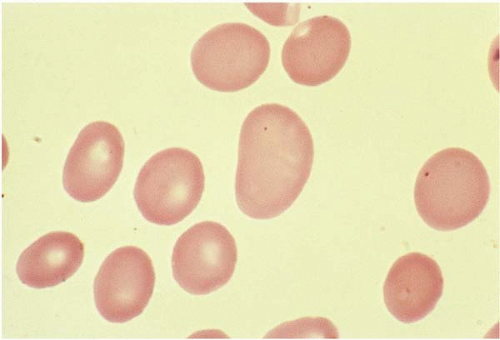 Figure 5.3 Macrocytes in the peripheral blood smear from a patient with vitamin B12 deficiency. A macro-ovalocyte is in the center of the field. |
Red Blood Cell Shape
Acanthocytes (spur cells) are RBCs with pointed membrane spicules of uneven length. The most common inherited condition is abetalipoproteinemia. Acquired conditions include liver disease, post-splenectomy, and malabsorption. In contrast to Burr cells, acantocytes cannot regain a normal shape.
Bite cells are semicircle remnants of RBCs after being partially phagocytosed or after extrusion of the Heinz body from the RBC. They are seen in G6PD deficiency, unstable hemoglobins, and oxidant hemolysis where the Heinz body is pitting by the spleen (Fig. 5.4).
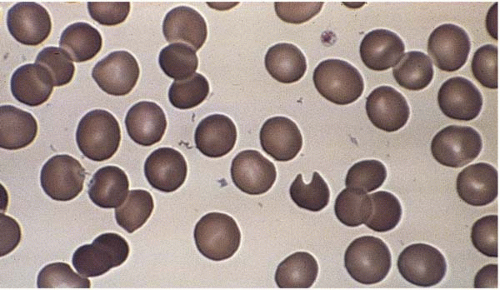 Figure 5.4 Normochromic normocytic RBC with a bite cell in the center of the field. |
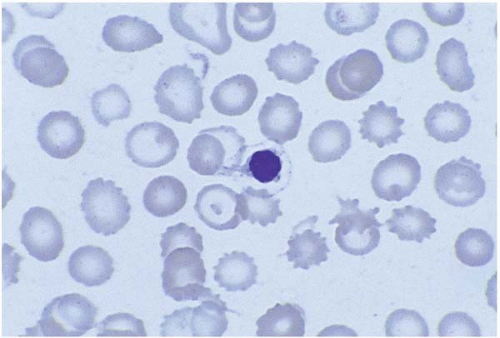 Figure 5.5 Peripheral blood smear contains Burr cells and a target cell in the center of the field. |
Burr cells are an acquired RBC membrane abnormality with short, evenly spaced spicules. They are seen in uremia, liver disease, burn, pyruvate kinase deficiency, gastric ulcer, or cancer, and are difficult to distinguish from echinocytes. The spicules of Burr cells may revert to normal (Fig. 5.5).
Codocytes (target cells) have a dense central Hb surrounded by a colorless ring due to membrane redundancy. The osmotic fragility is reduced. Codocytes are characteristic of thalassemia, hemoglobinopathy, postsplenectomy, liver disease, and iron-deficiency anemia (see Fig. 5.5).
Dacrocytes (teardrop cells) are drop-shaped RBCs seen in myelofibrosis and myelophthesis. They may also be seen in thalassemia, iron deficiency, and conditions where Heinz bodies are formed (Fig. 5.6).
Drepanocytes (sickle cells) are crescent-shaped RBCs with points at both ends as a result of hemoglobin polymerization in low-oxygen tension. They are typical of sickle cell disease. Upon oxygenation, most sickle cells revert to normal.
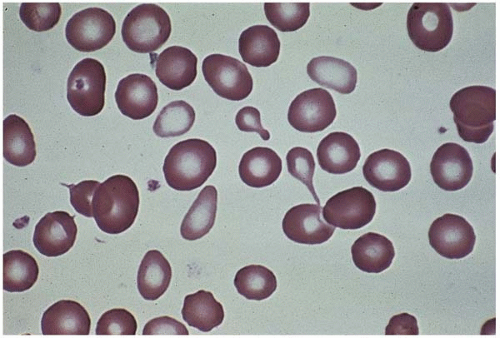 Figure 5.6 Peripheral blood smear contains occasional dacrocytes. |
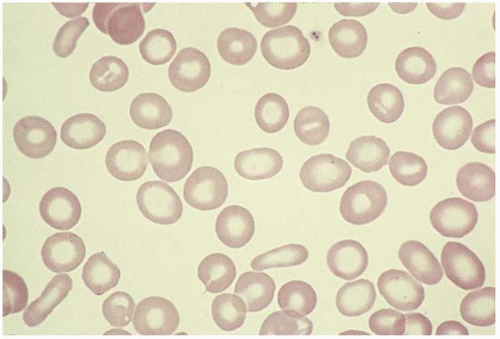 Figure 5.7 Peripheral blood smear contains occasional elliptocytes. |
Echinocytes (crenated cells) have uniform blunt membrane spicules and represent artifact. They are difficult to distinguish from Burr cells.
Elliptocytes are pencil-shaped RBCs similar to ovalocytes. They are most often seen in hereditary elliptocytosis (Fig. 5.7).
Ovalocytes are egg-shaped RBCs with varying hemoglobin content. Although the exact mechanism is unclear, an ovalocyte is highly variable and may appear normochromic or hypochromic, normocytic or macrocytic. They are often seen in megaloblastic anemia (see Fig. 5.3).
Schistocytes are RBC fragments due to mechanical injury, commonly seen in malignant hypertension, vasculitis, thrombotic thrombocytopenic purpura, hemolytic uremic syndrome, disseminated intravascular coagulation (DIC), and heart valve replacement. Membrane defects such as spherocytes and antibody-binding red cells may cause decreased cell survival, and therefore increased schistocytes (Fig. 5.8).
Spherocytes are small, round RBCs with a decreased surface-to-volume ratio, a smaller MCV, and no central pallor.
The MCHC is elevated (>36%). They are characteristic of hereditary spherocytosis, immune hemolytic anemia, and transfusion reaction (Fig. 5.9).
The MCHC is elevated (>36%). They are characteristic of hereditary spherocytosis, immune hemolytic anemia, and transfusion reaction (Fig. 5.9).
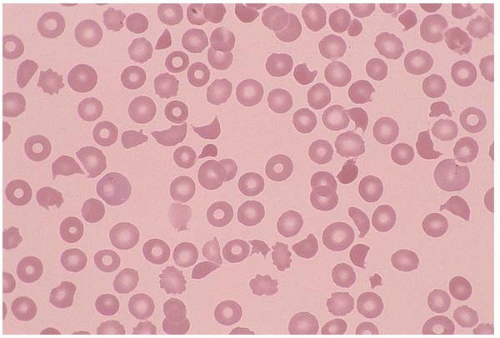 Figure 5.8 Peripheral blood smear contains schistocytes and increased reticulocytes in a patient with thrombotic thrombocytopenic purpura. |
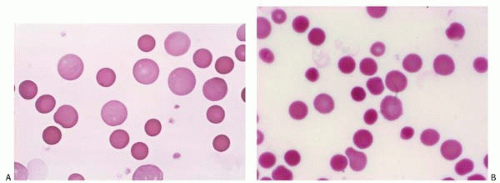 Figure 5.9 (A,B) Peripheral blood smear contains spherocytes in a patient with autoimmune hemolytic anemia. |
Stomatocytes are RBCs with a “slit-like” central pallor. They are seen in hereditary stomatocytosis, alcohol toxicity, liver disease, and Rh null phenotype. Some drugs (e.g., chlorpromazine and phenothiazine) may induce reversible stomatocytosis. More commonly, stomatocytes present as artifact (Fig. 5.10).
Red Cell Inclusions
Basophilic stippling represents ribosomes and mitochondria remnants distributed homogeneously over the cells. It is seen in arsenic and lead poisoning, thalassemia, megaloblastic anemia, alcoholism, and conditions causing accelerated heme synthesis (Fig. 5.11). Increased hemoglobin concentration and punctuate basophilia of red cells have also been seen in patients with chronic cyanide exposure.
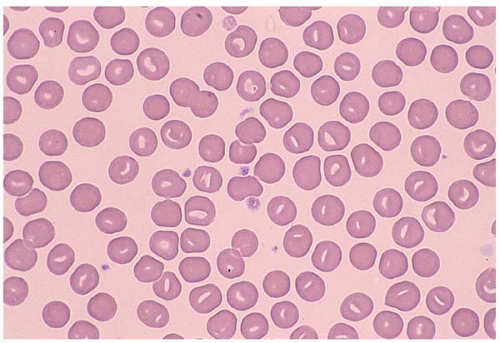 Figure 5.10 Peripheral blood smear contains stomatocytes in a patient with hereditary stomatocytosis. |
Cabot ring is a ring or figure-8-like structure appearing reddish blue on Romanowsky stain. It contains arginine-rich histone and non-heme iron, and represents mitotic spindles or microtubule remnants. It may be seen in megaloblastic anemia, hemolysis, overwhelming infection, lead poisoning, myelodysplasia, thalassemia, and postsplenectomy.
Heinz bodies are 0.3- to 2-μm denatured hemoglobin attached to red cell membrane, best visualized with crystal violet, brilliant cresyl blue, or methylene blue stains. They are seen in G6PD deficiency, α-thalassemia, unstable hemoglobinopathy syndromes, chemical insult, and oxidant stress. Multiple Heinz bodies give the RBCs a “golf ball” appearance.
Howell-Jolly bodies are nuclear remnants. Normally, the spleen pits these fragments effectively. During stress, the inclusion formation exceeds the splenic pitting mechanism and Howell-Jolly bodies appear. They are seen after splenectomy or in hemolytic anemia, thalassemia, megaloblastic anemia, and alcoholism (Fig. 5.12).
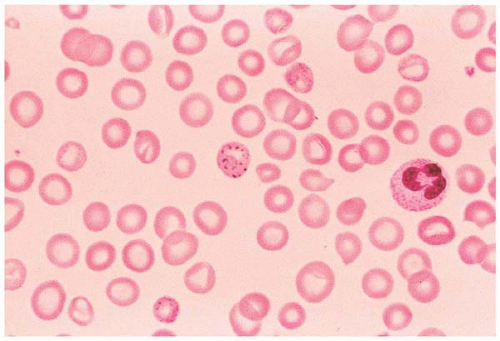 Figure 5.11 Peripheral blood smear from a patient with lead poisoning. The red cell in the center of the field shows basophilic stippling. |
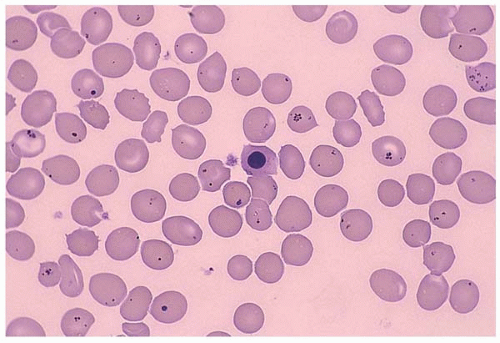 Figure 5.12 Peripheral blood smear contains many Howell-Jolly bodies in a postsplenectomy patient. |
Pappenheimer bodies (Siderotic granules) are non-heme iron at the cell periphery. Perl Prussian Blue stain reveals siderotic granules as small magenta inclusions. On Wright stain, they are called Pappenheimer bodies. Siderotic granules are seen in sideroblastic anemia, hemoglobinopathy, post-splenectomy, hemochromatosis, and hemosiderosis (Fig. 5.13).
Red Blood Cell Distribution
Agglutination appears as red cell clumping throughout the blood smear at room temperature. Agglutination persists with saline dilution but may disappear with sample warming. It occurs when an antibody against a specific antigen in the patient’s RBC antigen is present in his or her plasma. This may be seen in cold agglutinin disease or paroxysmal cold hemoglutinuria (Fig. 5.14).
Rouleaux formation results from elevated plasma globin. RBCs appear as stacks of coins. In contrast to agglutination, saline dilution disperses rouleaux. Rouleaux is common in multiple myeloma, Waldenstrom macroglobulinemia, chronic lymphocytic leukemia, and hyperproteinemia (Fig. 5.15).
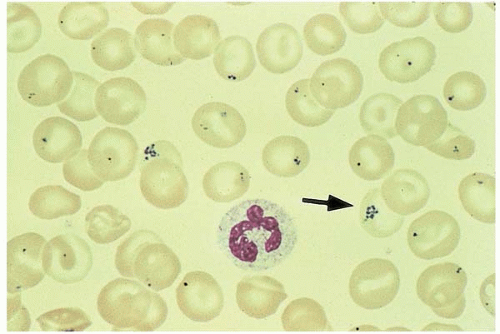 Figure 5.13 Peripheral blood smear contains many RBCs with Pappenheimer bodies (siderotic granules). |
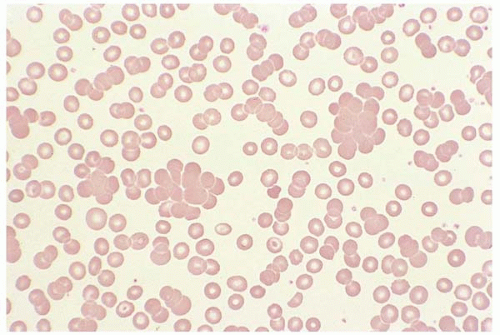 Figure 5.14 Peripheral blood smear shows RBC agglutination in a patient with cold agglutinin disease. |
HEMOLYTIC ANEMIA
Hemolytic anemia may be congenital or acquired. The congenital abnormalities include RBC membrane, enzymes, an Hb structure and synthesis, and are discussed in Chapter 4. Acquired hemolytic anemia is typically immune-mediated or microangiopathic; both are characterized by macrocytosis, polychromasia, and reticulocytosis. Autoimmune hemolytic anemia shows microspherocytosis and a positive Coombs’ test, or direct antiglobulin test. Microangiopathic hemolytic anemia shows peripheral red cell fragments (schistocytes), decreased serum haptoglobin level, and, in many cases, evidence of coagulopathy. The degree of erythropoiesis is commensurate with the degree of anemia and hemolysis. Concomitant megakaryocytic hyperplasia is
often present. Macrophages and erythrophagocytic activity may be increased. Dyserythropoiesis may be present, likely due to the stress of rapid erythrocyte production. Anemia develops when RBC destruction surpasses the production.
often present. Macrophages and erythrophagocytic activity may be increased. Dyserythropoiesis may be present, likely due to the stress of rapid erythrocyte production. Anemia develops when RBC destruction surpasses the production.
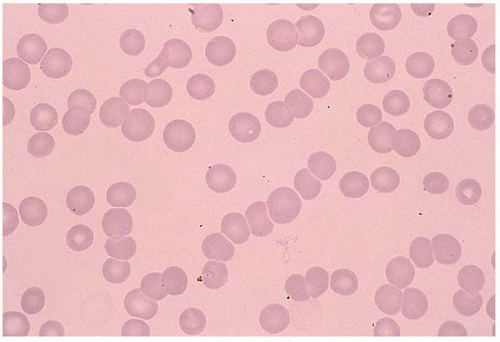 Figure 5.15 Peripheral blood smear shows Rouleaux formation. |
Hemolysis can be intravascular (7) or extravascular. Most hemolysis is extravascular due to phagocytosis by mononuclear phagocytic cells in the liver, spleen, and the bone marrow (8, 9, 10). Heme degradation produces bilirubin, which binds to albumin when entering the liver. In the liver, the water-insoluble bilirubin is converted to a soluble form and is excreted in the bile, which is then converted to urobilinogen by the intestinal bacteria and eliminated in the feces. Some urobilinogen is reabsorbed from the gut and excreted in the urine. Patients with extravascular hemolysis have jaundice, elevated bilirubin, and elevated urine and fecal urobilinogen.
In intravascular hemolysis, the released Hb binds to haptoglobin to form the Hb-haptoglobin complex, the latter is subsequently cleared by the liver. Therefore, a reduction in plasma haptoglobin indicates intravascular hemolysis. When the released Hb exceeds the Hb-binding capacity of heptoglobin, free Hb appears in the plasma to give rise to hemoglobinemia. Free Hb passes through renal glomeruli to cause hemoglobinuria. Heme from the circulating Hb binds hemopexin to form heme-hemopexin complex, which is then removed by the liver. When hemopexin is exhausted, hematin is formed and binds albumin to produce methemalbumin. Methemalbumin gives the plasma a brown color. Thus, in addition to reduced haptoglobin, other changes in intravascular hemolysis include reduced hemopexin, methemalbuminemia, hemoglobinemia, hemoglobinuria, and hemosiderinuria.
Immune Hemolytic Anemia
As the most common form of acquired hemolysis, immune hemolytic anemia can be classified as autoimmune, alloimmune, or drug-induced (11). Regardless of the etiology, its characteristic features are the presence of microspherocytes. Erythroid hyperplasia occurs in essentially all hemolytic disorders; bone marrow examination usually is not required.
Autoimmune Hemolytic Anemia
Autoimmune hemolytic anemia indicates premature RBC destruction by autoantibodies. It may be idiopathic, or secondary to malignancy, infection, graft-versus-host disease, and drugs, and can be induced by warm or cold antibodies.
Warm autoimmune hemolytic anemia accounts for up to 70% of autoimmune hemolytic anemia; most cases are extravascular and do not cause autoagglutination. It is usually caused by IgG, although IgM and IgA may be detected. Fc receptor-mediated immune adherence to RBC and compliment-mediated hemolysis are proposed mechanisms. The presence of compliment on the RBC facilitates IgG-mediated phagocytosis. The most useful test in diagnosing warm autoimmune hemolytic anemia is the direct autiglobulin test (Coombs’ test) using IgG, with or without compliment C3d. Warm autoantibody usually has specificity for Rh antigen, although other antigens have also been described. Spherocytosis in the peripheral blood indicates hemolysis. Marked reticulocytosis reflects marrow compensation for RBC loss. The indirect Coombs test may detect circulating antibodies when hemolysis is active. Coombs-negative autoimmune hemolytic anemia may be caused by a low RBC antibody titer or hemolytic anemia with an antibody other than IgG.
Corticosteroids and splenectomy are used in the treatment of warm autoimmune hemolytic anemia. Postsplenectomy patients should receive vaccination against bacteria S. pneumonea, meningococcus, and H. influenza.
Cold autoimmune hemolytic anemia (cold agglutinin syndrome) is a compliment-mediated hemolysis at temperature 10°C to 30°C, most commonly seen in the elderly. Cold autoantibodies are usually IgM, with 15% being biphasic IgG (Donath-Landsteiner antibody), especially in the pediatric group. The most common antibody in cold autoimmune hemolytic anemia is anti-I in mycoplasma pneumoniae and infectious mononucleosis.
RBC agglutinate at body extremities when the temperature is <30°C. The compliment is activated and hemolysis is more likely when thermal reactivity is >30°C. Occasionally, the blood film shows neutrophil-RBC rosettes. Coombs test is positive for C3d but negative for IgG. Pathologic anti-I antibody has a broad thermal range (0°C to 32°C) whereas normal anti-I in healthy individuals has a thermal range of 0°C to 22°C. This difference is significant because compliment is most hemolytic at 22°C or higher and pathologic anti-I is able to cause hemolysis. Examined at room temperature, the peripheral blood shows RBC agglutination (Fig. 5.16). At 37°C, the agglutination dissolves (Fig. 5.17).
Paroxysmal cold hemoglobinuria accounts for 35% to 40% of acute transient hemolytic anemia in children during viral infections (e.g., measles, mumps, influenza, and
chickenpox). Syphilis infection may also result in paroxysmal cold hemoglobinuria. It is caused by a cold biphasic IgG (Donath-Landsteiner antibody), mostly IgG3, against blood group P antigen. Antibody binds RBC at cold temperature in the peripheral circulation. The compliment is activated and intravascular hemolysis occurs when RBC returns to warmer temperature (37°C).
chickenpox). Syphilis infection may also result in paroxysmal cold hemoglobinuria. It is caused by a cold biphasic IgG (Donath-Landsteiner antibody), mostly IgG3, against blood group P antigen. Antibody binds RBC at cold temperature in the peripheral circulation. The compliment is activated and intravascular hemolysis occurs when RBC returns to warmer temperature (37°C).
 Figure 5.16 Scanning electron microscopy shows RBC agglutination. |
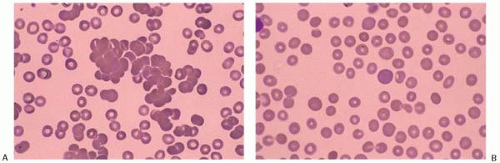 Figure 5.17 (A) Peripheral blood smear shows RBC agglutination (room temperature) in a patient with cold agglutinin disease. (B) Peripheral blood smear shows dissolution of RBC agglutination at 37°C in the same patient with cold agglutinin disease. Note the increased number of reticulocytes. |
Both direct and indirect antiglobulin tests are available. The indirect test has a higher sensitivity. The Coombs test is performed by incubating the sample at 0°C for 60 minutes, then at 37°C for 30 minutes. If Donath-Landsteiner antibody is present, it binds RBC at cold temperature and hemolysis occurs upon warming. The indirect test is performed by mixing the patient’s serum with P antigen-positive RBC, then adding fresh serum as a compliment source for hemolysis. Staying warm usually avoids hemolysis. Symptomatic patients may require transfusion.
Alloimmune Hemolytic Anemia
Alloimmune hemolytic anemia occurs when the immune system is sensitized to another individual’s RBC antigen and an antibody is formed. The most common forms are hemolytic transfusion reaction and hemolytic disease of the newborn.
Hemolytic Transfusion Reaction
The acute transfusion reaction accounts for more than 80% of hemolytic transfusion reactions; most are due to ABO incompatibility, usually caused by IgM (6,12). The blood film shows both agglutination and spherocytosis in massive hemolysis. If the antibody is not previously present, a less severe, delayed hemolytic reaction occurs in 7 to 14 days. Although most patients were previously alloimmunized, this antibody becomes undetectable over time, and the only evidence may be a positive Coombs. The antibody in delayed reaction is usually IgG against Rh, Duffy, Kidd, and hemolysis is extravascular.
Hemolytic disease of the newborn is also called erythroblastosis fetalis. During pregnancy, the maternal IgG is transferred to the fetus through placenta. Hemolysis occurs when there is blood group incompatibility between the mother and the fetus. Antibodies against Rh, ABO, and Kell are the major causes, with anti-D being the most severe. This usually occurs in the second and subsequent pregnancy. Infants are anemic and jaundiced with hepatospenomegaly. Severely affected infants have kernicterus. Stillbirth may occur. Rh-negative women should receive Rh immunoglobulin at 28 weeks gestation and within 72 hours of exposure to D+ RBC. Kleihauer-Betke test or flow cytometry analysis is useful in determining the amount of fetal cells in maternal blood so the amount of Rh immunoglobulin can be adjusted.
Drug-Induced Hemolytic Anemia
Drug-induced hemolysis may be immune-mediated or due to oxidative injury. The former can be intravascular hemolysis or extravascular hemolysis, and is usually caused by one of the three mechanisms: autoantibody production (e.g., α[asymptotically equal to]methyldopa) where the antibody directs against Rh antigen in many patients (this is similar to warm autoimmune hemolytic anemia); hapten-dependent antibody formation (e.g., penicillin); or formation of antibody-protein complex (e.g., quinine, quinidine, and rifampin). Hemolytic anemia is usually reversible upon discontinuation of the drug. Coombs test is positive in many patients and may persist for up to two years after hemolysis resolves. The strength of the oxidant, its blood level, the presence of G6PD deficiency, and glutathione-dependent pathways determine the oxidative injury. Older RBCs are more prone to oxidative injury. The characteristic features of oxidative hemolysis include
the formation of methemoglobin, sulfhemoglobin, and Heinz bodies.
the formation of methemoglobin, sulfhemoglobin, and Heinz bodies.
Drugs likely causing oxidative hemolysis include nitrofurantoin, dapsone, aminosalicytic acid and, rarely, high-dose oxygen treatment in vitamin E deficiency. Demonstration of drug-dependent hemagglutination in Coombs test confirms the diagnosis.
Nonimmune Hemolytic Anemia
Infection
Infection-induced hemolysis may be caused by malaria, C. perfringen, and toxoplasma gondii or other microorganisms (Fig. 5.18). Microorganisms injure RBC through different mechanisms including physical invasion, hemolysin secretion, antibody formation, and infection-associated DIC. Antibiotics may also cause hemolysis. In some cases, multiple mechanisms coexist.
Mechanical Injury
Mechanical injury includes large-vessel hemolytic anemia (malignant hypertension, prosthetic heart valves) and microangiopathic hemolytic anemia as seen in thrombotic thrombocytopenic purpura, hemolytic uremic syndrome, DIC, autoimmune vasculitis, and march hemoglobinuria. The peripheral blood smear usually contains Burr cells and schistocytes.
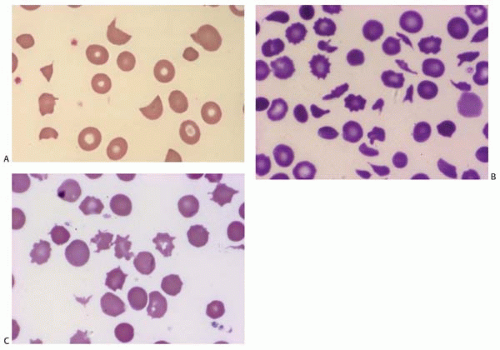 Figure 5.18 (A) Peripheral blood smear contains schistocytes in a patient with meningococcal septicemia. (B) Peripheral blood smear contains schistocytes in a patient with cardiac valve disease. (C) Peripheral blood smear contains schistocytes, Burr cells, and macrocytes in a patient with alcohol liver disease and Zieve syndrome. |
Thermal and Osmotic Injury
Red cells are very fragile to high-temperature exposure and changes in osmotic pressure. Burn results in fragmentation and membrane loss of RBC with formation of microspherocytes, schistocytes, and Burr cells. Hemoglobinemia and hemoglobinuria occur the first day after a burn. The morphologic damage is self-limiting. Freshwater or saltwater drowning causes hemolysis due to abrupt osmotic change in pulmonary circulation.
Other Causes
Patients with Wilson disease may present with acute hemolytic anemia and fulminate hepatic failure (13). Prompt diagnosis reduces the high mortality associated with this entity. Hemolytic anemia associated with vitamin E deficiency has been reported in infants with failure to thrive. Patients with chronic alcoholic liver disease may
develop severe hemolytic anemia with circulating burr cells (echinocytes) (14). Echinocytosis in these cases are said to result from an alteration in the membrane phosphatidylserine and phosphatidylinositol concentrations. Zieve syndrome is manifested by hyperlipemia, hemolytic anemia and alcoholic fatty liver (15). Although hemochromatosis is most likely the cause of the erythrocyte anomaly, alcohol intake is probably responsible for the onset of hemolytic anemia. Phytosterolemia (sitosterolemia) is a recessively inherited metabolic condition in which cholesterol and plant-derived cholesterol-like molecules are absorbed unselectively. Hematological presentation includes stomatocytic hemolysis, macrothrombocytopenia (Mediterranean stomatocytosis’macrothrombocytopenia) and elevated levels of phytosterols in the blood (16). Other reported cases associated with hemolytic anemia include industrial toxins such as arsine.
develop severe hemolytic anemia with circulating burr cells (echinocytes) (14). Echinocytosis in these cases are said to result from an alteration in the membrane phosphatidylserine and phosphatidylinositol concentrations. Zieve syndrome is manifested by hyperlipemia, hemolytic anemia and alcoholic fatty liver (15). Although hemochromatosis is most likely the cause of the erythrocyte anomaly, alcohol intake is probably responsible for the onset of hemolytic anemia. Phytosterolemia (sitosterolemia) is a recessively inherited metabolic condition in which cholesterol and plant-derived cholesterol-like molecules are absorbed unselectively. Hematological presentation includes stomatocytic hemolysis, macrothrombocytopenia (Mediterranean stomatocytosis’macrothrombocytopenia) and elevated levels of phytosterols in the blood (16). Other reported cases associated with hemolytic anemia include industrial toxins such as arsine.
Paroxysmal Nocturnal Hemoglobinuria
Paroxysmal nocturnal hemoglobinuria (PNH) is a rare acquired form of intravascular hemolysis caused by an abnormal clone of pleuripotent stem cells with sensitivity to compliment. All hematopoietic cell lines are affected (17).
More than 20 proteins are missing from the red cell surface in patients with PNH, with glycophosphatidylinositol (GPI) being the key glycolipid in anchoring the proteins to RBC membrane. The defect of GPI synthesis is caused by mutation of the pig-A gene on chromosome Xp22.1. Deficiency of the delay accelerating factor (DAF, CD55), homologous restriction factor (HRF), or membrane inhibitor of reactive lysis (MIRL, CD59) accelerates compliment complex and gives rise to episodic hemolysis, particularly at night when the pH is decreased (18). Three types of compliment sensitivity were detected in PNH: RBCs in PNH I react normally in the presence of compliment, whereas PNH II cells are three to five times more susceptible to lysis; PNH III cells have 20 times or higher susceptibility to lysis. The level of deficiency provides information on the disease severity.
The peripheral blood shows pancytopenia and a lower leukocyte alkaline phosphatase activity in the neutrophils. The bone marrow is hypoplastic despite significant hemolysis. Storage iron is deficient. Sucrose hemolysis test is the screening test, with more than 5% RBC lysis indicating PNH. Ham test is confirmatory: When the compliment fixes the RBC at a decreased pH, RBC from the PNH patients lyse, whereas normal RBCs are resistant to lysis. Flow cytometry analysis using CD55 and CD59 provides a sensitive and specific measure of hemolysis, and helps subdivide the abnormalities into partial or complete deficiency. Cytogenetic analysis demonstrates loss of chromosome Y and trisomy 9 in some patients.
Patients with PNH may progress to aplastic anemia or acute myelogenous leukemia (19), and are complicated with thrombotic episodes at uncommon sites.
REVIEW OF IRON METABOLISM
The Hb molecule consists of iron, protoporphyrin IX, and globin. Deficiency of any of them results in Hb deficiency. Three major mechanisms are involved in abnormal iron metabolism: decreased iron supply (iron-deficiency anemia); defective recycling of storage iron (anemia of chronic disease); and defective utilization (sideroblastic anemia, lead poisoning).
Food derived from animals and plants provide sufficient iron. Iron absorption depends on its supply, storage, and demand. A healthy man has 1 to 2 mg absorbed iron. Dietary iron in ferric form (Fe+++) is converted to ferrous form (Fe++) in the stomach for absorption. In the blood, ferrous iron is reconverted to ferric form again by ferroxidase and is transported to transferrin. Once bound to tranferrin, iron is delivered to tissue and organs freely. In the liver, iron is released from transferrin to form ferritin, the storage form. Hemosiderin is another form of storage iron in macrophage lysosomes after ferritin level is reached. Hemosiderin is typically visualized by Perl stain.
Each milliliter of RBC production requires 1 mg of iron. A healthy adult loses 20 to 25 mL of RBC daily (1% of red cells); therefore, 20 to 25 mg of iron is needed daily. Of these, 5% (1 to 2 mg) comes from dietary supply, and 95% comes from recycling of senescent RBC. This recycling recovers essentially all iron except that lost in feces, urine, sweat, desquamation, and menstruation.
MICROCYTOSIS AND MICROCYTIC ANEMIA
Microcytic anemia is defined as the presence of small, often hypochromatic RBCs in the peripheral blood smear and is characterized by a low MCV (<80 fL). Despite abundant iron supply, iron-deficiency anemia is the most common cause of microcytic anemia in the world. In adults, anemia of chronic disease is probably more common. The differential diagnosis includes thalassemia minor, hemoglobinopathy, and sideroblastic anemia.
The diagnosis of iron deficiency in the elderly is difficult due to the common presence of chronic diseases that are associated with a high ferritin level. Recent studies suggested transferrin receptor-ferritin index to be a more sensitive iron measurement, and is able to diagnose iron-deficiency anemia with a higher sensitivity (88%) (20, 21, 22, 23). In microcytic or normocytic anemia, a bone marrow examination is still the gold standard in distinguishing iron-deficiency anemia from non-iron-deficiency anemia (24, 25, 26, 27, 28, 29, 30, 31, 32).
The main differential diagnosis of microcytosis includes, in decreasing frequency, iron deficiency anemia, thalassemia minor, inflammation, or chronic renal failure. In patients with normocytic anemia, disseminated malignancy and acute blood loss should always be considered (Table 5.1) (33,34).
TABLE 5.1 Differential Diagnosis of Microcytosis and Microcytic Anemia | |||||
|---|---|---|---|---|---|
|
Iron-Deficiency Anemia
Iron-deficiency anemia results from insufficient storage iron. It may be caused by inadequate intake, increased demand, or increased loss (35). Clinical manifestations range from asymptomatic, mild, to severe. The process can be divided into three stages.
Stage 1 (iron depletion) starts when storage iron is utilized. Serum ferritin and marrow hemosiderin are decreased. In response, iron absorption from the gastrointestinal mucosa is increased. The peripheral blood findings including a normal RBC and reticulocyte count, and a normal serum iron level. A persistent negative balance leads to iron exhaustion and the development of iron-deficient erythropoiesis.
Stage 2 (iron deficient erythropoiesis) begins when serum iron level and transferrin saturation start to fall. Serum transferrin (to increase iron absorption) and free erythrocyte protoporphyrin (FEP) are elevated as a result of erythroid hyperplasia. The RBC number and morphology remain normal.
Stage 3 (iron-deficiency anemia) is characterized by hypochromic microcytic anemia. Peripheral blood shows decreased Hb, MCV, MCHC, and an elevated RDW. RBCs are hypochromic microcytic with poikilocytosis (elliptocytes, ovalocytes, and folded forms) (Fig. 5.19). Reticulocyte count is typically normal or slightly elevated. Platelet count is often elevated. The bone marrow biopsy shows ineffective erythropoiesis with small normoblasts containing scanty cytoplasm and ragged cytoplasmic boarders. The dysplastic changes of the erythroid series are not prominent in iron-deficiency patients (Fig. 5.20). Perl stain reveals absent storage iron. When bleeding is the primary cause, leukocytesis and thrombocytosis may occur.
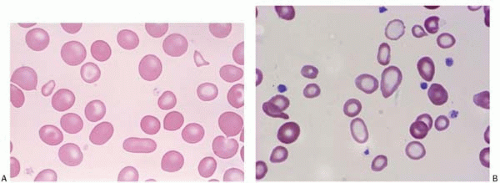 Figure 5.19 (A) Peripheral blood smear shows hypochromic microcytic anemia in a patient with iron-deficiency anemia. (B) Peripheral blood smear shows hypochromic microcytic anemia in a patient with iron-deficiency anemia and lead poisoning. |
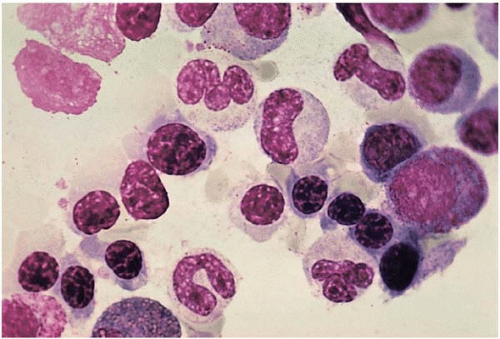 Figure 5.20 Bone marrow aspirate smear shows ineffective erythropoiesis with normoblasts containing ragged cytoplasmic boarders in an iron-deficient patient. |
The classic signs of iron-deficiency anemia include pallor, glossitis, koilonychias, and systolic heart murmur. Pica and angular stomatitis are common. Without treatment, patients may reveal decreased myeloperoxidase or T-cell activity, although these deficits are difficult to measure. An iron panel study (serum iron, ferritin, total iron binding capacity [TIBC], and tranferrin saturation) helps provide a specific diagnosis of iron-deficiency anemia: serum iron, ferritin and iron saturation are decreased, TIBC is elevated.
The main differential diagnosis includes constitutional disorders of iron metabolism and defective iron reutilization, such as in anemia of chronic disease; the latter shows hypochromic microcytic anemia, low serum iron, low to normal TIBC, and increased storage iron in the marrow.
Identifying the underlying cause and providing iron supplement are the mainstay of treatment. The reticulocyte count begins to rise in a few days and reaches a maximum in 7 to 10 days. The Hb usually normalizes in 2 months.
Sideroblastic Anemia
Sideroblastic anemia is a diverse group of hypochromic disorders with defective iron utilization within erythropoietic cells. It is characterized by a basic triad of hypochromic anemia, ringed sideroblasts, and hyperferremia. Consequently, hemoglobin synthesis is impaired and surplus iron collects in mitochondria. This imbalanced cellular iron uptake and its impaired incorporation into heme becomes more prominent during the late stages of erythroid maturation. Thus, siderotic rings are most conspicuous in orthochromatic erythroblasts. The iron-containing mitochondria often form a ring around the nucleus to give the pathognomonic “ringed sideroblast” (Fig. 5.21). These ringed sideroblasts are unsuitable for circulation and expire within the marrow, thus the term “ineffective erythropoiesis.” Meanwhile, the accumulation of defective sideroblasts generates the impression of erythroid hyperplasia.
Three criteria should be met before sideroblasts are properly diagnosed: (a) the iron-staining granules must be abnormally large; (b) the granules must exceed five to six per cell; and (c) they must form a perinuclear arc extending around one-third or more of the nucleus. The peripheral blood shows hypochromic microcytic red cells with dimorphism in most of the cases. Siderocytes are seen in some patients.
Sideroblastic anemias may be hereditary or acquired. The hereditary forms are less common, among which the X-linked sideroblastic anemia is a better described entity. Molecular genetic abnormalities include mutations in δ-aminolavulinic acid S2 (ALAS2) gene and genes involved in ataxia, thiamine-responsive megaloblastic anemia, and Pearson marrow-pancreas syndrome (36).
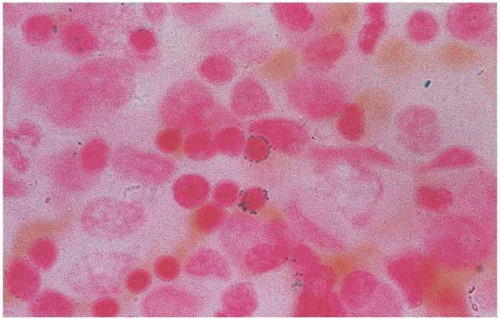 Figure 5.21 Bone marrow aspirate smear shows ringed sideroblasts in a patient with zinc-induced copper deficiency |
Acquired forms result from a variety of causes, including alcoholism, copper deficiency (37, 38, 39, 40, 41, 42, 43, 44), drugs (45, 46, 47), infection (48), and myelodysplastic syndrome. The pathogenesis in most cases of acquired sideroblastic anemia is unknown.
In alcohol-induced sideroblastic anemia, the differential diagnosis includes megaloblastic anemia, myelodysplastic syndrome, and acute leukemia. Patients with copper deficiency may have ringed sideroblasts and cytoplasmic vacuolization in the erythroid and myeloid precursors (49, 50, 51, 52) (Fig. 5.22). Zinc supplement (e.g., used in patients with acne) causes copper deficiency by competing with copper for gastrointestinal absorption and subsequent sideroblastic anemia (53). Zinc toxicity may present with anemia, leukopenia, and cytoplasmic vacuolization of myeloid and erythroid precursors (54, 55, 56). Copper-deficient infants due to limited diet may respond to copper supplement with a brisk increase in neutrophils and reticulocyte count. Certain drugs (e.g., chloramphenical, isoniazide, fusidic acid, alkylating agents, and immunosuppressants) cause sideroblastic anemia by interfering with ALA-S or heme synthase activity (57,58); anemia usually resolves after discontinuation of the responsible drugs. Rare causes of sideroblastic anemia include pregnancy (59) and hypothermia (60).
Idiopathic sideroblastic anemia is a clonal disorder in adults and the elderly, with ringed sideroblasts >15%. By French-American-British and World Health Organization (FAB and WHO) classifications, it corresponds to myelodysplastic syndrome, refractory anemia with ringed sideroblasts (MDS’RARS). Chromosome abnormalities are detected in up to 50% of the patients, with chromosomes 5, 7, 8, 20, and Y being the most frequently affected.
Laboratory test suggests iron overload with elevated transferrin saturation. Ringed sideroblasts usually disappear
within 2 weeks after stopping alcohol intake. Pyridoxine supplement may be helpful (Table 5.2).
within 2 weeks after stopping alcohol intake. Pyridoxine supplement may be helpful (Table 5.2).
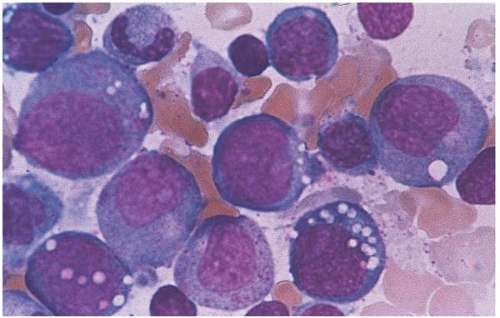 Figure 5.22 Bone marrow aspirate smear shows erythroid and myeloid precursors with cytoplasmic vacuoles. The patient has sideroblastic anemia.
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|