This section discusses reactive changes in monocytes and histiocytes, as well as constitutional and acquired storage disorders, which are predominantly manifested as histiocytic hypertrophy and hyperplasia. Clonal disorders of monocytes and histiocytes are discussed in later chapters.
MONOCYTES
Monocytopenia
Peripheral blood monocytopenia is an uncommon finding, diagnosed when the absolute monocyte count is less than the normal range of the testing laboratory. In our laboratory, the lower limit of normal is 0.200 × 109‘L.
Monocytopenia occurs in many of the same settings as neutropenia, especially in bone marrow failure (Table 8.1). In postchemotherapy clinical settings, monocytopenia is a powerful predictor of neutropenia and life-threatening infection, especially with mycobacteria and fungi. It is not always clear whether monocytopenia is a risk factor for infection or a sequela of infection. This is particularly true for infections acquired in the course of another disease associated with monocytopenia, such as hairy cell leukemia.
Monocytopenia may be a variable finding in the individual patient (1, 2, 3). Monocytopenia may be episodic, apparently of autoimmune origin; or cyclic, in synchrony with cyclic neutropenia.
Monocytopenia is associated with drug therapy, including cancer chemotherapy (4,5), cladribine (6,7), corticosteroids (8), imatinib mesylate (9), and leflunomide (10). It has also been reported to accompany alcohol abuse (11), aplastic anemia (12), late-stage chronic lymphocytic leukemia (13), hairy cell leukemia (13, 14, 15), hemodialysis (16,17), lymphopenia (18,19), rheumatoid arthritis (20), and severe burns (21).
Monocytopenia is a feature of infection by human immunodeficiency virus (22,23) and Salmonella typhi (24), and is found in severe acute respiratory syndrome (SARS) (25) and tuberculosis (26,27).
Monocyte apoptosis has been reported as an effect of therapy with thalidomide and infliximab (anti-tumor necrosis factor) (28,29).
The differential diagnosis includes constitutional (familial) monocytopenia (30).
Reactive Monocytosis
Peripheral blood monocytosis is diagnosed when the absolute monocyte count exceeds the normal range of the testing laboratory. In our laboratory, the upper limit of normal for individuals 6 years old and above is 0.950 × 109‘L. Infants and children up to 6 years old have a higher upper limit; in this age range, individual laboratory values should be consulted.
The clinical settings for reactive monocytosis are diverse (Table 8.2) (31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53). They include acute myocardial infarction; exercise; therapy with granulocyte-macrophage colony-stimulating factor and granulocyte colony-stimulating factor; malaria, syphilis, tuberculosis, and other infectious diseases; major depression; obstructive airway disease; tissue damage and trauma; myelodysplastic syndrome; Hodgkin lymphoma; hairy cell leukemia; adult T-cell leukemia’lymphoma; and disseminated carcinoma. We have also noted reactive monocytosis in the bone marrow in Diamond-Blackfan anemia.
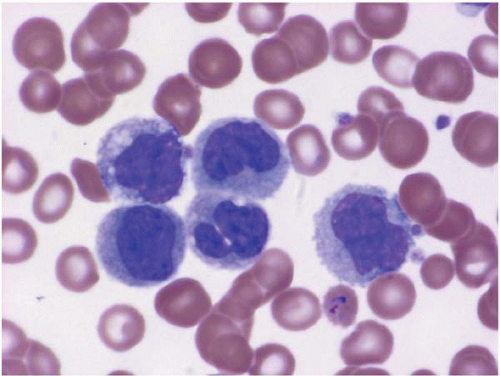
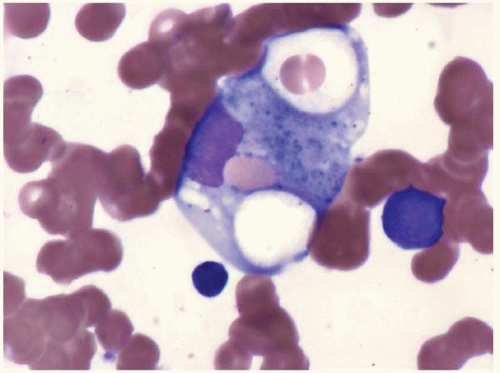
The peripheral blood smear shows increased monocytes, which may include mature and activated forms containing cytoplasmic vacuoles (Figs. 8.1 and 8.2). Reactive monocytosis may be accompanied by reactive eosinophilia and’or basophilia.
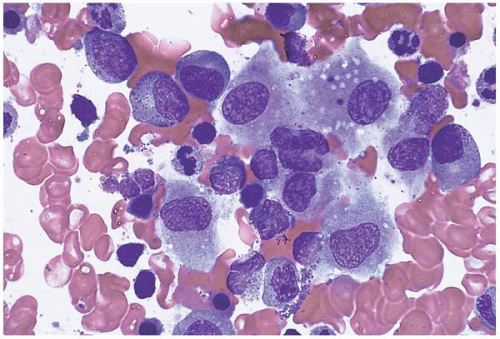
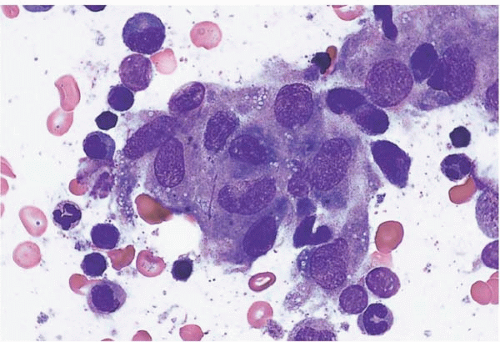
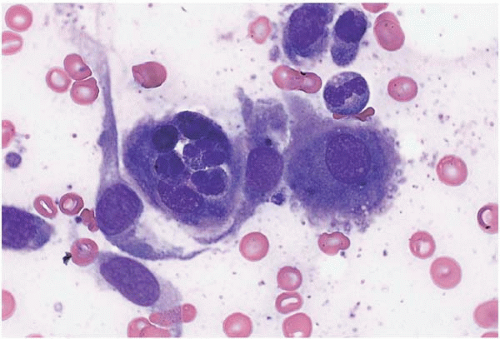
Bone marrow aspirate smears show increased monocytes, monocytic precursors, and histiocytes, which may be accompanied by increased eosinophils, basophils, and’or mast cells (Figs. 8.3). Granuloma-like clusters of histiocytes may be present (Fig. 8.4).
Histologic sections of the bone marrow may appear less cellular than normal because of the increased number of monocytes and histiocytes with abundant, pale-staining cytoplasm. Granulomas may be present. Hemophagocytosis may be present, and is discussed more fully below.
TABLE 8.1 Conditions Associated with Monocytopenia
Alcohol abuse
Aplastic anemia
Chronic lymphocytic leukemia, late stage
Drug therapy
Hairy cell leukemia
Hemodialysis
Non-human immunodeficiency-related lymphopenia
Rheumatoid arthritis
Severe burns
Human immunodeficiency virus infection
Salmonella typhi infection (typhoid fever)
Severe acute respiratory syndrome (SARS)
Tuberculosis
The differential diagnosis of reactive monocytosis in the peripheral blood consists primarily of atypical lymphocytosis due to viral infection, especially with automated differential counts; and clonal monocytic disorders.
Monocytic Leukemoid Reaction
Monocytic leukemoid reaction has been described in congenital syphilis and in a patient treated with granulocyte colony-stimulating factor (G-CSF) for acute myeloid leukemia (AML) (54, 55, 56, 57). Monocytosis remitted upon withdrawal of G-CSF therapy. Careful consideration should be given to such cases to exclude acute monocytic leukemia.
Figure 8.1 Reactive monocytosis, peripheral blood. This specimen is from a patient withPlasmodium vivaxmalaria; an intraerythrocytic organism is present.
TABLE 8.2 Conditions Associated with Reactive Monocytosis
Acute Myocardial Infarction
Adult T-cell leukemia’lymphoma
Atypical mycobacteriosis
Brucellosis
Diamond-Blackfan anemia
Disseminated carcinoma
Exercise
Granulocyte and granulocyte-macrophage colony-stimulating factor therapy
Hodgkin lymphoma
Infectious disease
Leishmaniasis
Major Depression
Malaria
Myelodysplastic Syndrome
Parvovirus B19 infection
Syphilis
Tissue damage
Trauma
Varicella virus infection
Histone deacetylase inhibitor valproic acid (VPA) was recently shown to enhance proliferation and self-renewal of normal hematopoietic stem cells, raising the possibility that VPA may also support growth of leukemic progenitor cells (LPC) (58). Here, VPA maintains a significantly higher proportion of CD34+ LPC and colony-forming units compared to control cultures in six AML samples, but selectively reduces leukemic cell numbers in another AML sample with expression of AML1’ETO. Our data suggest a differential effect of VPA on the small population of AML progenitor cells and the bulk of aberrantly differentiated blasts in the majority of AML samples tested.
Figure 8.2 Reactive monocytosis with vacuolation, peripheral blood. Two monocytes with cytoplasmic vacuoles are present in this specimen from a patient with hairy cell leukemia.
Figure 8.3 Reactive histiocytosis, bone marrow aspirate. Several histiocytes are present showing abundant pale, basophilic, slightly granulated cytoplasm in this specimen from a patient with Hodgkin disease.
HISTIOCYTES
Reactive Histiocytosis
Both the number and phagocytic activity of histiocytes are increased in a wide variety of settings, distinguished primarily by increased and’or ineffective hematopoiesis (59, 60, 61, 62, 63). These include megaloblastic anemia, leptospirosis, hemoglobinopathies, human immunodeficiency virus infection, immune-mediated cytopenias, sarcoidosis, vasculitis, and myelodysplastic syndrome.
Figure 8.4 Reactive histiocytosis, bone marrow aspirate. A cluster of reactive histiocytes, consistent with a granuloma, is shown in this specimen from a patient with Hodgkin lymphoma.
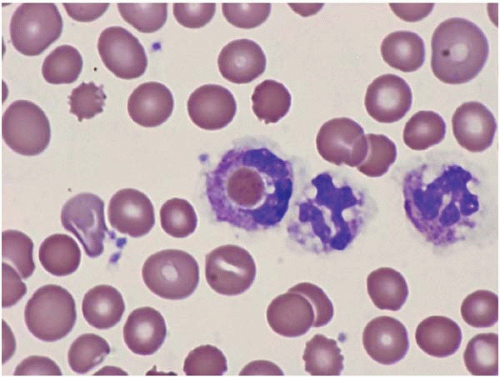
Figure 8.5 Erythrophagocytosis, peripheral blood. A neutrophil with an engulfed erythrocyte is present in a patient with myelodysplastic syndrome, who did not have evidence of hemophagocytic syndrome.
The peripheral blood may show evidence of hemophagocytosis (Fig. 8.5).
Bone marrow aspirate smears show an increased number of morphologically benign histiocytes containing ingested hematopoietic cells, nuclear remnants, and’or blue-green granules (Figs. 8.6 and 8.7).
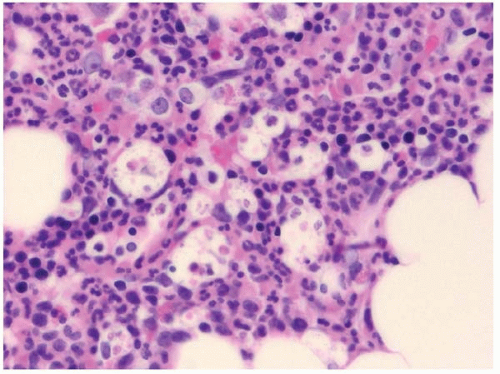
Histologic sections of the bone marrow show increased histiocytes scattered throughout the hematopoietic tissue (Figs. 8.8 and 8.9). They are apparent on hematoxylin and eosin staining primarily because of their abundant pale, retracted cytoplasm, which causes apparently “empty” spaces among the hematopoietic cells.
Figure 8.6 Erythrophagocytosis, bone marrow aspirate. This cell was found in a staging bone marrow examination for Hodgkin lymphoma in an asymptomatic patient.
Figure 8.7 Reactive histiocytosis with hemophagocytosis, bone marrow aspirate. The patient had Hodgkin lymphoma without systemic manifestations of hemophagocytic syndrome.
No systemic signs or symptoms are associated with this type of reactive histiocytosis, in contrast to the findings in hemophagocytic syndrome.
Hemophagocytic Syndrome
Hemophagocytic syndrome (HPS, also known as macrophage activation syndrome) occurs sporadically and as a familial disorder, termed hemophagocytic lymphohistiocytosis (FHLH). FHLH is discussed separately below.
HPS is a life-threatening condition in which normal histiocytes, under the influence of excessive cytokine stimulation, proliferate and exhibit extreme hemophagocytosis (Table 8.3) (64).
Figure 8.8 Reactive histiocytosis, bone marrow biopsy. This patient had follicular lymphoma without evidence of hemophagocytic syndrome. Low power.
Figure 8.9 Reactive histiocytosis, bone marrow biopsy. This patient had follicular lymphoma without evidence of hemophagocytic syndrome.
TABLE 8.3 Conditions Associated with Hemophagocytic Syndrome
Constitutional Syndromes
Chédiak-Higashi syndrome
Griscelli syndrome
Hermansky-Pudlak syndrome
Lysinuric protein intolerance
Veno-occlusive disease with immunodeficiency
X-linked lymphoproliferative disease
X-linked osteopetrosis, anhydrotic ectodermal dysplasia, and immunodeficiency
Acquired Nonneoplastic Conditions
Young age
Old age
Pregnancy
Asplenism
Ethylene glycol poisoning
Immune-mediated thrombocytopenia
Inflammatory bowel disease
Juvenile rheumatoid arthritis
Kikuchi disease (necrotizing lymphadenitis)
Mosquito bite allergy
Polyarteritis nodosa
Rheumatoid arthritis
Systemic lupus erythematosus
Solid organ transplantation
Neoplastic Conditions
B-cell neoplasms
Hodgkin lymphoma
T-cell neoplasms
NK- and T’NK-cell neoplasms
Langerhans cell histiocytosis
Carcinoma
HPS in general requires two factors: a background of immune dysfunction, especially affecting cytotoxic T cells and natural killer (NK) cells; and exposure to an infection, drug, or toxin as the triggering event. HPS has rarely been described in apparently normal, healthy individuals, but an underlying genetic or acquired disorder cannot be excluded in such patients (65, 66, 67).
Immune dysfunction predisposing to HPS occurs in certain constitutional syndromes, and in acquired conditions and diseases with an immune-mediated component.
Constitutional syndromes tend to develop HPS in infancy or childhood as an accelerated, often terminal, phase of disease. In such cases, HPS is usually triggered by Epstein-Barr virus (EBV) infection. These syndromes include Chédiak-Higashi syndrome, Griscelli syndrome, and Hermansky-Pudlak syndrome, further discussed in Chapter 4 (68, 69, 70). Other syndromes predisposing to HPS include lysinuric protein intolerance, veno-occlusive disease with immunodeficiency (VODI), X-linked lymphoproliferative disease, and X-linked osteopetrosis, anhydrotic ectodermal dysplasia, and immunodeficiency (XL-O-EDA-ID) (71, 72, 73, 74).
Acquired conditions and diseases predisposing to HPS are numerous, and have in common an aspect of impaired immunity or disturbed immune status (Figs. 8.10 and 8.11; see Table 8.5). The very young, the elderly (75), and pregnant women (76,77) appear to be at risk, as well as those without splenic function (78). Diseases in which HPS has been reported include immune-mediated thrombocytopenia (79), inflammatory bowel disease (80), juvenile rheumatoid arthritis (81), Kikuchi disease (82,83), mosquito bite allergy (84), polyarteritis nodosa (85), systemic lupus erythematosus (77,86), rheumatoid arthritis (87), solid organ transplantation (88,89), B-cell neoplasms (76,90, 91, 92, 93), Hodgkin lymphoma (94), T-cell neoplasms (90,95,96), NK- and T’NK-cell neoplasms (90,97,98), Langerhans cell histiocytosis (99), carcinoma (100), and ethylene glycol poisoning (101).
HPS caused by Epstein-Barr virus (EBV) may provoke a clonal T-cell proliferation, and it may be difficult in such cases to determine whether hematolymphoid neoplasia is a predisposing factor or a consequence of HPS (102,103).
Infections reported with HPS are caused by a very broad array of organisms, both common pathogens and opportunistic and unusual organisms. All types of infectious agents have been reported to trigger HPS, including bacteria, mycobacteria, viruses, fungi, and parasites. The most frequently reported infectious agent is EBV (104, 105, 106, 107). EBV has been implicated not only as a trigger for HPS, but as an agent in the subsequent evolution of HPS to clonal T-cell disease (102,103)
Laboratory studies in HPS show a characteristic pattern of pancytopenia, abnormal liver function tests, hypertriglyceridemia, hyperferritinemia, coagulopathy, and increased serum levels of cytokines.
Patients may present at any age. Those with preexisting constitutional syndromes tend to develop HPS as a terminal event in childhood. The principal clinical features are fever, wasting, and hepatosplenomegaly. The peripheral blood shows cytopenias. Circulating hemophagocytic macrophages have been reported (108).
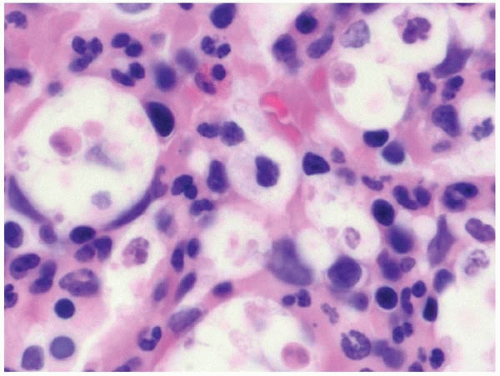
Bone marrow aspirate smears show variable numbers of histiocytes containing ingested hematopoietic cells (Fig. 8.10) (109). In infants, few histiocytes and little or no phagocytosis may be seen, and biopsy of the liver or another site may be required for diagnosis. Histologic sections of the bone marrow show increased histiocytes with abundant, eosinophilic, faintly granular cytoplasm and ingested nuclei. Iron stains may be helpful in identifying histiocytes. Interstitial T-cells may be increased. Hematopoietic precursors may show myelodysplastic changes, possibly related to increased cell turnover (110,111).
HPS may mask an underlying malignancy, especially acute leukemia or malignant lymphoma (Fig. 8.11). Thus, a specific and careful search for tumor cells is essential in evaluating bone marrow specimens with HPS.
The differential diagnosis of HPS includes sepsis; acute lymphoblastic leukemia (due to HPS-induced lymphoproliferation); and hemophagocytosis due to myelodysplastic syndrome, acute myeloid leukemia, and malignant histiocytosis (112, 113, 114, 115, 116). In cases of myeloid neoplasia and malignant histiocytosis, the cells showing hemophagocytosis are malignant, not reactive, and are cytologically and histologically abnormal.
Only gold members can continue reading. Log In or Register to continue